1. アルツハイマー病(Alzheimer's disease)
アルツハイマー病(AD)の脳にはアミロイドβの細胞外蓄積(amyloid plaque, 老人斑)とリン酸化タウの神経細胞内蓄積(神経原線維変化; neurofibrillary tangles, NFT)の2つの病態が存在する。タウ病変はpretangle stageを含めると20歳代からはじまる加齢的な側面をもった変化であり、脳梗塞や頭部外傷などはこの病態を促進すると考えられる。アミロイドβの存在が直接的に神経細胞死を招くのではなく、タウ病変を誘発してNFTを引き起こすと推測されている(アミロイドカスケ-ド仮説)。糖尿病や高血圧(動脈硬化)はAD発症の危険因子とされ、アミロイドβの蓄積との関連性が指摘されている。近年の生体でのバイオマーカー測定技術の発展により、認知症発症前からADの病理の存在を検出できるようになった。
優性遺伝する家族性AD(全ADの1%程度)の遺伝子異常のほとんどがアミロイドβの産生にかかわる遺伝子であることから、病理で脳アミロイドβの蓄積(老人斑)が目立たなければADは否定的となる。老人斑は初期にはびまん性老人斑(diffuse plaque)のことが多く、進行とともに典型的な老人斑が増えてくる。AD脳では老人斑は前頭葉や側頭葉底面、次いで頭頂-側頭-後頭葉の連合野に出現し、辺縁系に出現するのはもっと病期が進行してからである。また、認知症を発症していない加齢脳とAD脳では老人斑の出現の程度に差はあっても明らかな境界はない。ADの画像検査としてアミロイドPETは病理的側面を裏付ける意味において診断に有用であるが、現時点(2021年)では保険診療として認められていない。替わりとして髄液のAβ42の減少やリン酸化タウの増加などを参考にするが、髄液採取には時間と手間がかかり、採血よりも侵襲は大きいので一般外来での実施は難しい。ADと診断するためには脳のアミロイドβの蓄積を確認することが必須であるが、脳のアミロイドβの蓄積が必ずしも認知症(タウ病変の広範な進展)をきたすとは限らないことに留意する必要がある。
アルツハイマー病の病理所見
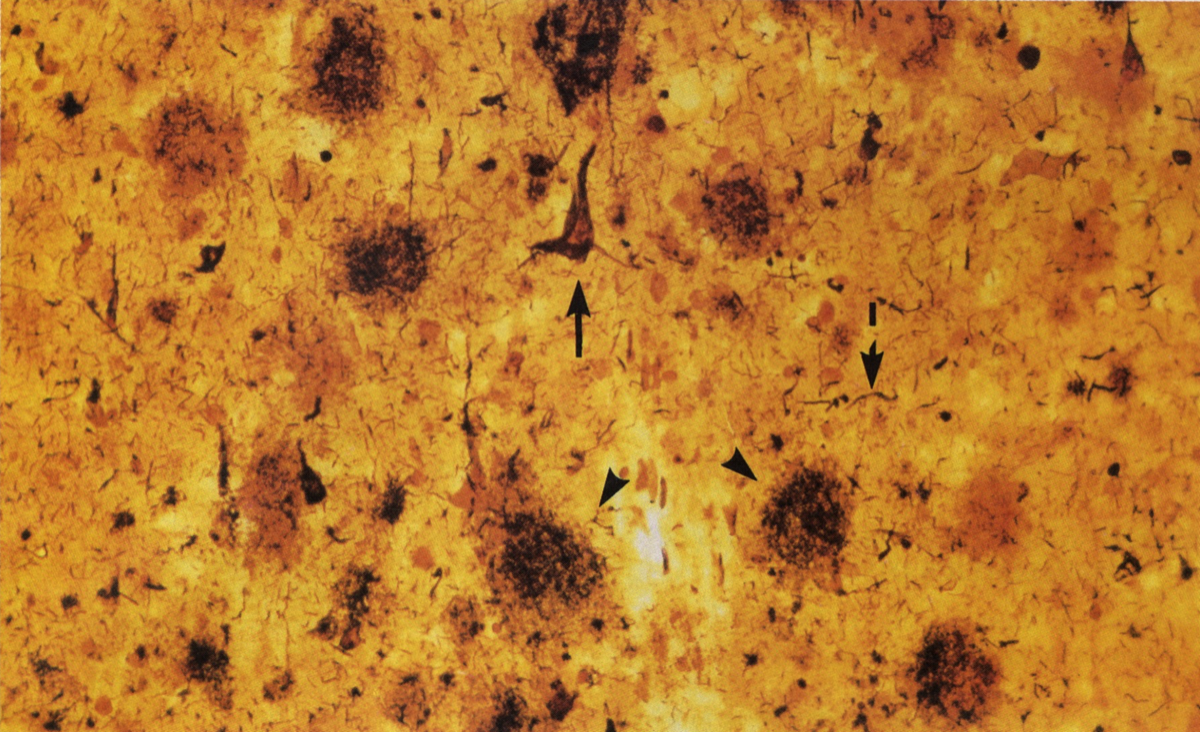
Amyloid plaque(矢頭)、neurofibrillary tangle (矢印)、neuropil threads (破線)(Yankner BA. et al., NEJM, 1991){1345415}
疫学
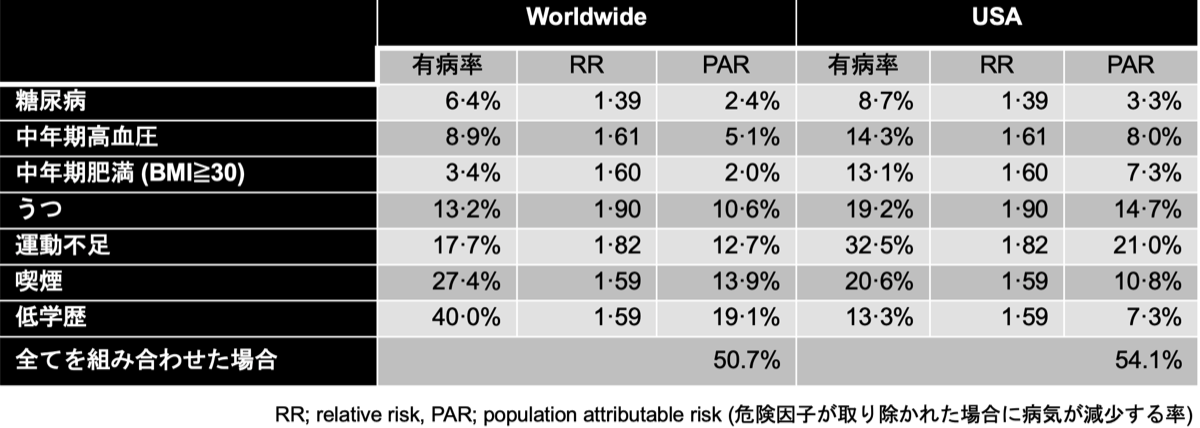
最近の欧米諸国の研究では、過去数十年間で有病率が20%〜60%、発症率が20%〜44%低下したという報告が相次いでいる。スウェーデンの研究の過去10年間のデータを解析した結果では、男性の有病率が6割低下した。英国の研究の過去20年間のデータを解析した結果、有病率は男性が4割、女性が2割低下した。これに対して、日本におけるADの有病率は増加傾向にあり、2012年の厚生労働省の報告によると65歳以上の15%前後(460万人)が認知症で、ADに限るとその有病率は10%程度(300万人)と考えられている。男女比については女性に多いと言われている。なお、2019年現在、日本の65歳以上は3千500万人、全人口の28%となっている。
ADの発症リスクを低下させる因子としては、糖尿病や高血圧、肥満などの治療介入が挙げられている(表){21775213}。
症状
ADの臨床経過は、ほぼBraakのNFT stageに一致する。ごく初期の段階(Transentorhinal stage)では臨床的に明らかな症状はないが、アミロイドβが脳に蓄積すると、そこからNFTは拡散し発症する。ADにおける病初期の中核症状は大脳辺縁系の障害による経験記憶の低下である (Limbic stage)。MRIでは嗅内野、海馬、側頭葉の萎縮が認められる。脳血流SPECTやFDG PET(保険適応外)では側頭葉から頭頂葉、後部帯状回から楔前回など、脳の後半部の血流低下(=代謝の低下)が認められる。これは辺縁系から投射先領域への神経活動の低下を反映していると思われるが、MRIのVBM解析でしばしばこれらの領域の脳萎縮が検出されることから、これらの領域の神経細胞の変性もあると思われる(後述のタウ病変のニューロン-ニューロン伝播を参照)。若年発症(65歳以前)の場合は海馬の萎縮よりも皮質の萎縮が目立つ症例が存在する{16904912}{18698140}。この傾向は優性遺伝するADの症例に多く見られることから、アミロイドβなどのタウ病変促進因子が発症の早期化に関係していると考えられる。
タウによる神経原線維変化(NFT)は脳萎縮と相関しており、MR画像における脳萎縮は間接的にタウ病変を見ていることになる(脳萎縮の全てが必ずしもタウ病変ではない)。海馬など辺縁系の萎縮はタウ病態以外でも認められるが、SPECTやFDG PETで脳後半部の代謝異常の有無はADとその他の認知症との鑑別に有用である。MRI検査でもVBM解析をすることにより、海馬以外の領域の脳萎縮の存在を客観的に評価できる。著者らはVBM解析ソフトであるBAADを開発しているが、解析に人工知能を用いて脳全体の脳萎縮のパターンから”ADらしさ”を数値として示すことにより、診断の補助に役立てないか検討している。
図にADの際に一般的に出現するNFT(脳萎縮)の領域とそれに伴う症状を示す。
BraakのNFTステージ(Ⅰ〜Ⅱ)では臨床的に軽度認知障害(MCI)の段階で、患者本人が経験記憶の低下を自覚していることが多い。この段階でADに進展しない症例は約50%であり、アミロイドβの蓄積がないSD-NFTやPARTの可能性がある。アミロイドβが蓄積しているとADに進展するリスクが高くなるが、観察期間や寿命にもよるが必ずしも全ての症例がADに進展するものではない。
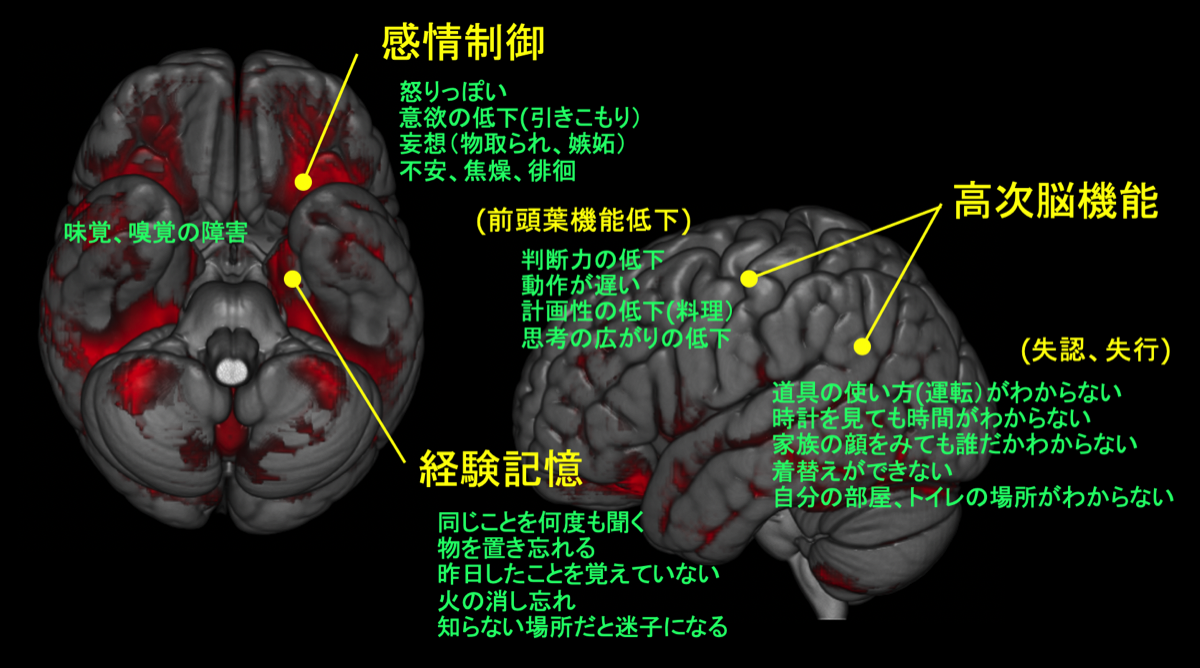
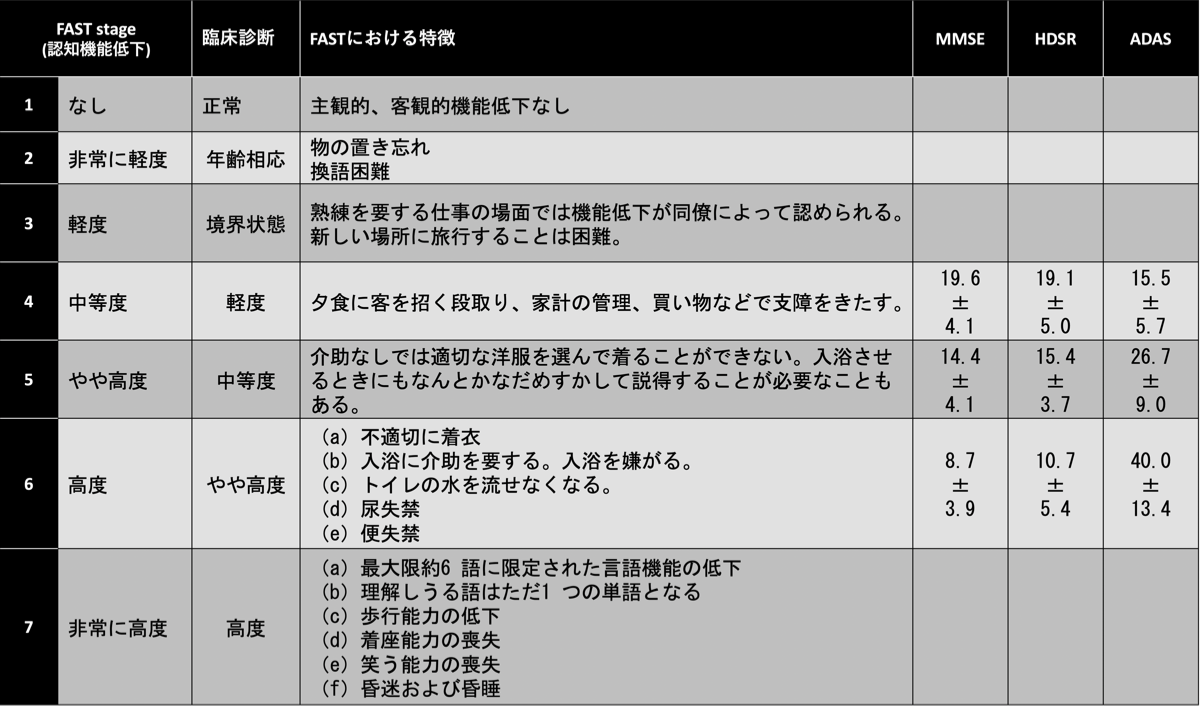
ADに進行すると新しく経験した記憶がなくなることから記憶力の低下を自覚しなくなる。同じことを何度も家族に聞く、物をどこに置いたか思い出せない、物をしまった場所が思い出せない、日常と異なる状況になると(例えば親戚の葬式、旅行先など)、記憶力の低下による障害が目立ち家族に気づかれることが多い。外来で最近の出来事を質問すると思い出せないためか、振り向いて家族にどうであったか聞いたりする(振り返り現象)。話の内容を取り繕うため、家族がいないと本人の話している内容が本当のことかどうかわからないことがしばしばある。例えば、趣味で何かをしていると答えても、家族に聞くと以前にはしていたが数年前から全くできていないことがわかる。薬の管理も同様で、本人はできているように話すが家族のサポートがないと服薬忘れが多い。BPSD(Behavioral Psychological Symptoms of Dementia;周辺症状)がなければ本人はいたって呑気で外来ではニコニコしていることが多いが、家族に忘れていることを指摘されると、(本人には自覚がないために)否定的な態度になり怒り出すことがよくある。最近の記憶はなく、以前の記憶は冗長に話したりする。病識がないため、デイサービスなどを勧めても行きたがらないことがある。さらに進行して、病変が側頭葉や頭頂葉など大脳皮質の後半部に及ぶと高次脳機能の障害(失行、失認、失語など)が目立つようになる。知らない道で迷う、自転車で出かけたのに徒歩で帰ってくるなどもよくある。また前頭葉の機能低下にともない、料理がうまくできない、料理の内容が乏しくなる、買い物に計画性がなくなるなどの症状が出てくる。判断力の低下から騙されやすくなったり、失敗を人のせいにしたり、感情のコントロ-ルができなくなったりする。判断力の低下は経験記憶の低下とあいまって、嫉妬妄想や物盗られ妄想を誘発しやすくなる。物を盗られたと勘違いして、自分で警察に連絡してトラブルになることがある。さらに進行すると時間、場所、人に対する見当識障害が出てくる。トイレの後始末ができなくなり、失禁をくり返すようになり、極端に活動性が低下してくる。MCIを含めてADの病期はFAST(functional assessment staging)でstage 1〜7まで分類される。ADの約80%にBPSDが出現すると言われている。
診断
受診時患者は自覚的に健忘症状があるため一人で来院する場合や、自覚症状はないが家族に付き添われて来院する場合など様々であるが、初診時においてはまず認知症かどうかを調べる必要がある。現病歴などで明らかに認知症の症状がある場合はMMSE検査などの簡易検査で十分なことも多いが、自覚的健忘を訴える症例や現病歴でどちらかわからない場合には、心理検査バッテリーによる詳しい認知機能評価が参考となる。ADの場合は経験記憶の障害が主体で、暗算や即時記憶よりも遅延再生(しばらくしてから記憶したことを思い出す)の障害が主体となる。初期の頃には、見当識障害はあまり目立たない。
入室時の観察は認知症のタイプ分類に役立つ。AD患者の場合は来室時に戸惑った様子(病識がないため)があっても、表情は普通で関節痛や腰痛がなければ動作もスムーズである。無表情で動作が遅く、歩行も不安定な場合は血管性認知症やレビー小体病の可能性がある。本人、家族とも表情が硬い場合にはBPSD(周辺症状)や前頭側頭葉タイプの認知症を念頭に診断を進める。歩行が小刻みでも開脚気味の場合には、血管性認知症や正常圧水頭症の可能性があり、腕振りが少ない場合や線を跨ぐ際にはスムーズな場合にはレビー小体病(パーキンソン病)の可能性がある。
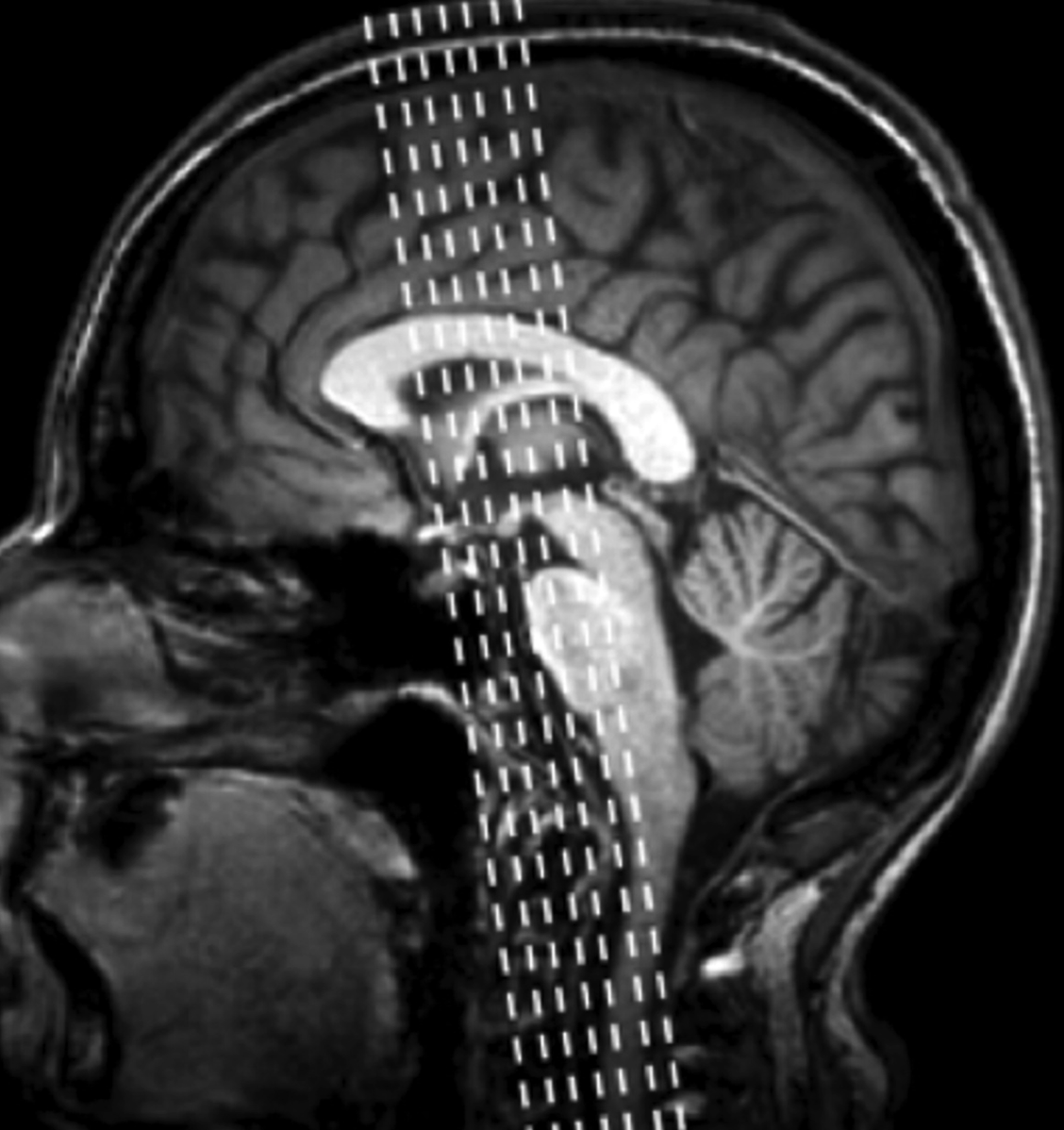
画像検査としては、はじめにMRIなどで脳梗塞や出血、腫瘍性病変などの鑑別をしておく必要がある。この際、VBM用の撮像を指示しておくことが望ましい。VSRADによる側頭葉内側(主に海馬)の萎縮の存在、BAADの人工知能(AI)によるADS(AD スコアー)や側頭葉の底面や外側の萎縮、後部帯状回の萎縮など萎縮のパターンを参考にする。SPECT検査で、側頭葉から頭頂葉、後部帯状回の血流低下が認められる場合にはADの確率が高い。
NINCDS/ADRDA研究班によるADの診断基準(2007年)
Probable AD:主要症状Aと一つ以上のB,C,DあるいはEの指示所見が必要。 A. 早期の有意な経験記憶障害がある
主要診断基準
1. 自覚・他覚的な6か月以上の緩徐進行性の記憶障害
2. 客観的検査による有意な経験記憶障害
3. 経験記憶障害のみか、進行に伴った他の領域の認知障害
支持する所見
B. 内側側頭葉の萎縮(海馬、嗅内皮質、扁桃体)
C. 髄液のアミロイドβ低下、総タウかリン酸化タウの増加
D. PET(FDGでADに特徴的な低下、アミロイドβの沈着)
E. 遺伝子変異の存在
除外基準
病歴(急性発症、早期からの歩行障害、痙攣、行動異常)
臨床症状(局所の神経症状、早期の錐体外路症状)
他の記憶障害を呈する疾患の除外(AD以外の認知症、大うつ病、脳血管障害、中毒、代謝障害)
Definite AD
臨床症状と脳病理所見が明らか(NIA-Reagan病理診断基準)
臨床症状と遺伝子変異が明らか
この診断による剖検脳との一致率は87.6%で、脳血管障害の合併は32%、パーキンソン病の合併は23%に認められた. {24849862}
International Working Group (IWG)-2 criteria for typical AD (2014) {24849862}
以下のAとBを満たす 6ヶ月以上にわたり、ゆっくりと進行する記憶障害が患者本人または介護者によって認められる 客観的な海馬由来の健忘症候群であり、これが記憶力と手がかり再生検査といったADに特異的かつ確立された経験記憶検査に基づいている
A. 特徴的な臨床的症状
早期から出現する有意な経験記憶の障害(単独でなくてもMCIや認知症を示唆するその他の認知障害あるいは行動異常が付随してもよい)があり以下の条件を満たす。
B. 生体検査でADの病理学的証拠として以下の項目から1つ
- 髄液のAβ1-42の減少とT-tauまたはP-tauの増加
- アミロイドPET陽性
- 優性遺伝する遺伝子異常の存在(PSEN1、PSEN2、APP)
現病歴
突然発症
以下の症状が早期から出現:歩行障害、痙攣、行動異常
臨床所見
局所の神経学的脱落所見
早期からの錐体外路徴候
早期からの幻覚
変動する認知機能レベル
その他の関連疾患
AD以外の認知症
大うつ病
脳血管障害
中毒、炎症、代謝障害
MRIのFLAIRまたはT2画像で側頭葉内側に感染や血管障害に矛盾しない信号変化の所見
認知機能検査
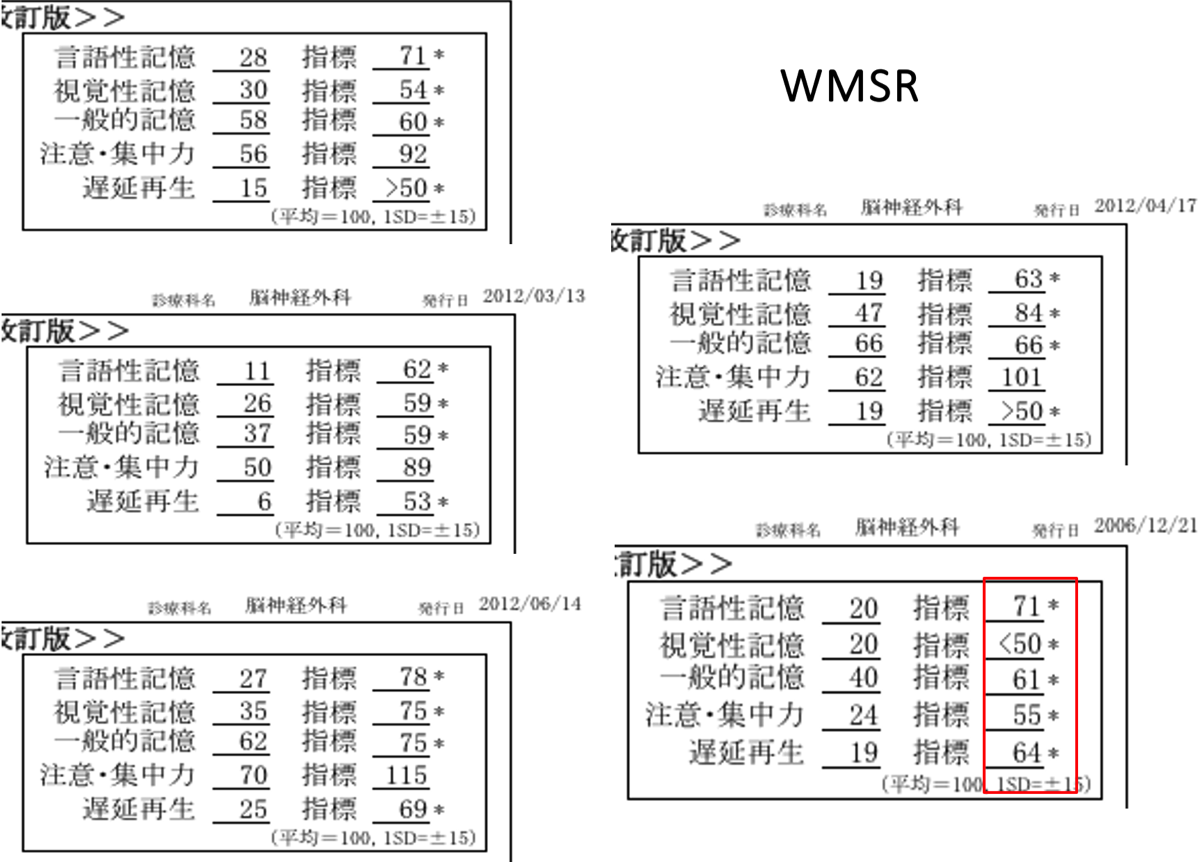
AD初期の認知機能評価では、遅延再生課題が重要となる。遅延再生課題とは、MMSEでは3つの単語あるいは品物を一旦記憶してもらい、別の課題をした後にもう一度思い出してもらう試験である。ウェクスラーメモリースケール(WMSR)の場合、年齢ごとに正常の平均値と標準偏差があるため、客観的な評価がし易い。ADの初期やMCIでは、遅延再生のみが1.5標準偏差を超えて低下していることが多い。
AD以外の場合(右図の右下)、注意集中の低下が目立ったり、視覚性記憶の低下のみが目立つことがある。特にレビー小体病では、注意集中の機能に変動が見られたり、図形問題で低い点数をとることが多い。
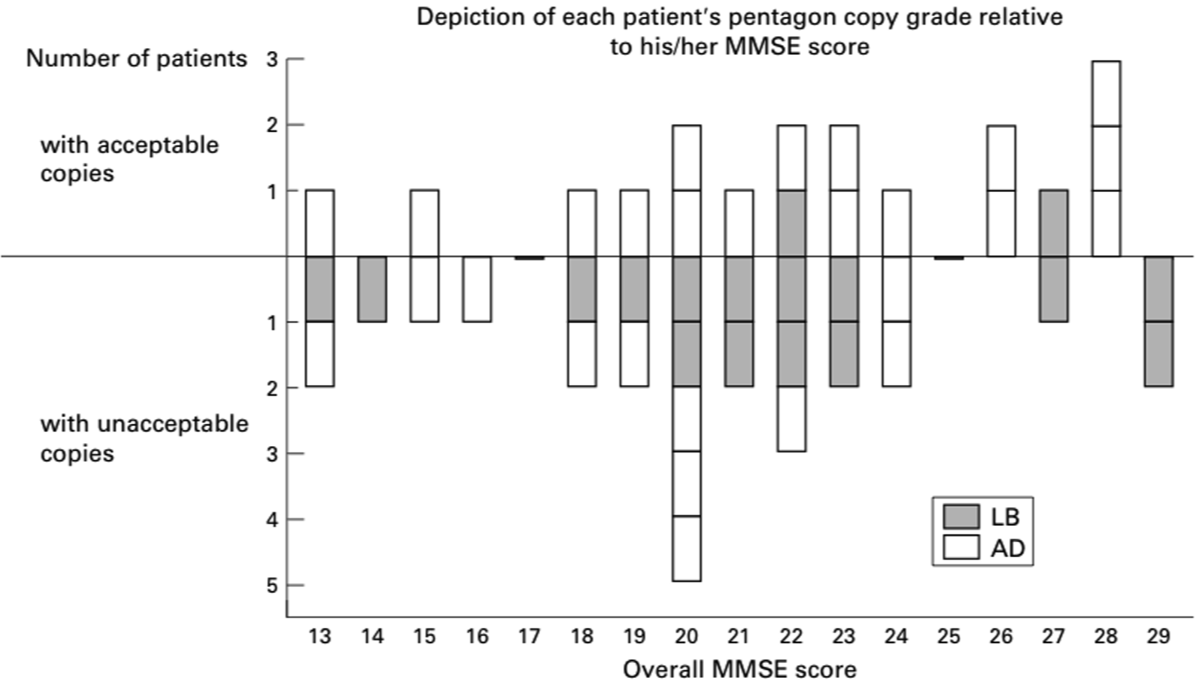
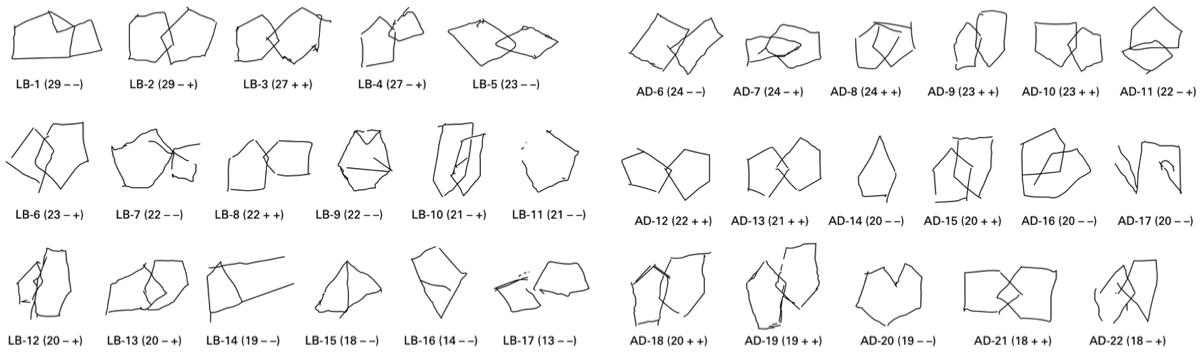
⇦MMSEにおける五角形の模写。ADに比べLBDの方が、この課題ができない割合が多い。(Ala T, et al., J Neurol Neurosurgeon Psychiatry, 2001) {11254771}
NFTのステージ分類
(Swarbrick S et al., Mol Neurobiol, 2019)
アミロイドβのステージ分類
画像検査
側頭葉内側、特に海馬の萎縮はAD患者に多いと言われている。レビー小体病(LBD)の患者よりもADでは海馬の萎縮が顕著である。しかしながら、最近の研究ではTDP-43単独でも海馬の萎縮を引き起こすことが明らかになりつつあり、「AD→海馬の萎縮」に関する特異性に問題があることがわかっている。またADとの混合型の認知症も多く認められる。MRIはADの直接的なバイオマーカーではないので、スクリーニング的な検査となる。
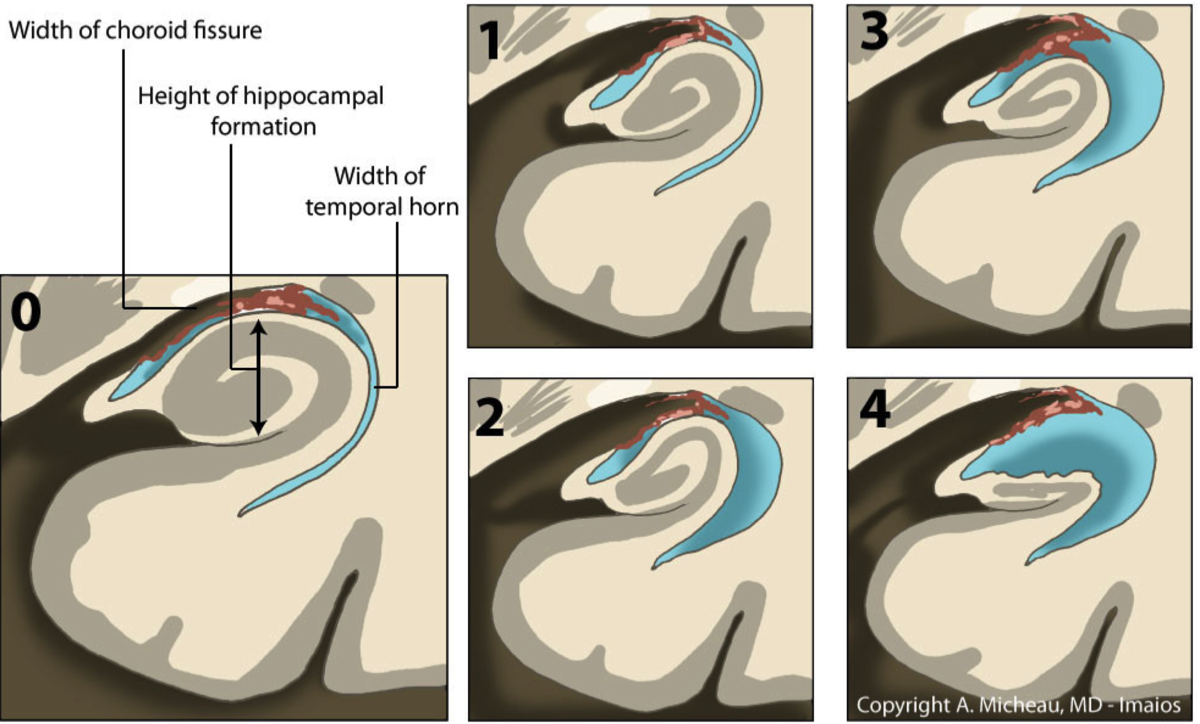
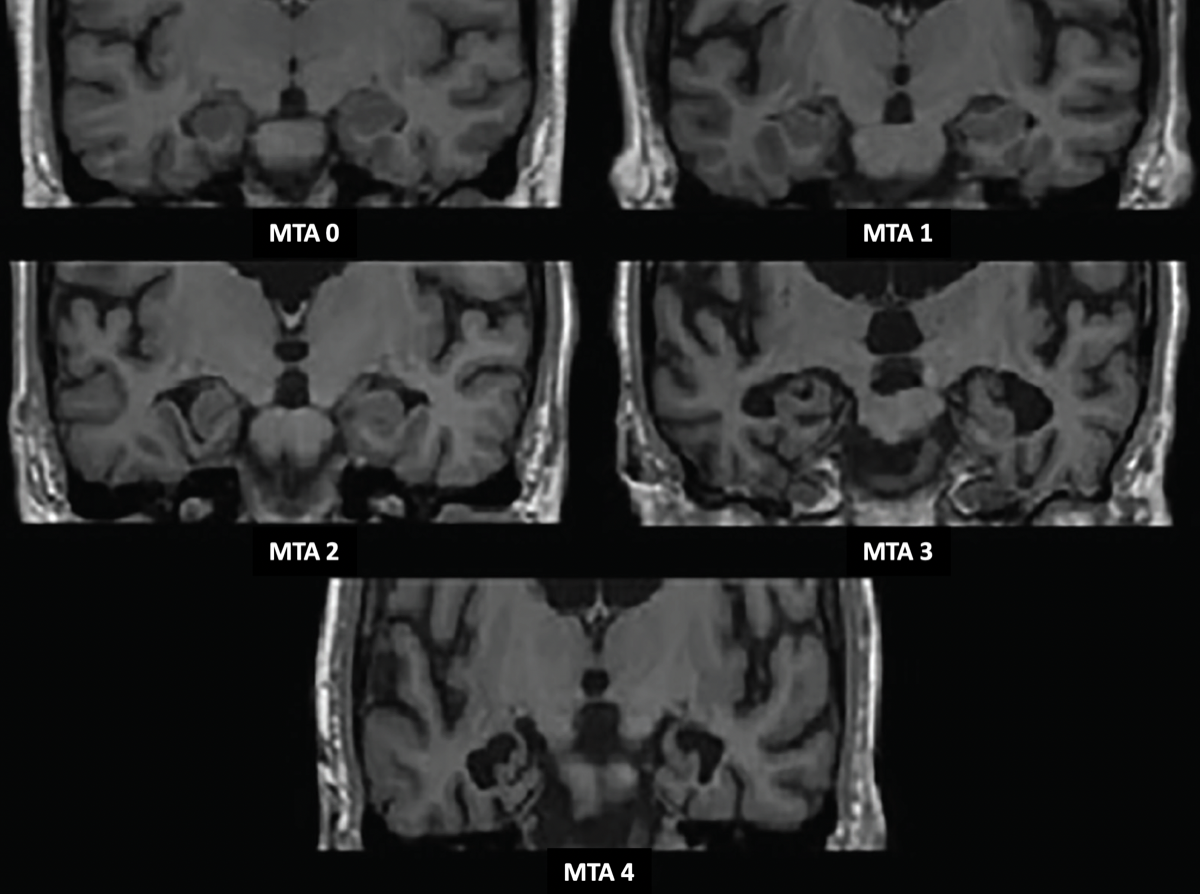
海馬の萎縮はMRIの冠状断で評価することが多く、視覚的なスケール評価法(Medial temporal lobe atrophy score)が報告されている。
MTA score (Scheltens' scale)の場合、75歳未満ではMTA2以上、75歳異常ではMTA3以上が有意な萎縮となる。
スコア 0:萎縮なし
スコア 1 : 脈絡裂の拡大のみ
スコア 2:側脳室の側頭角の拡大も見られる
スコア 3:海馬の体積減少(高さの減少)が中程度
スコア 4 : 海馬の重度の体積減少
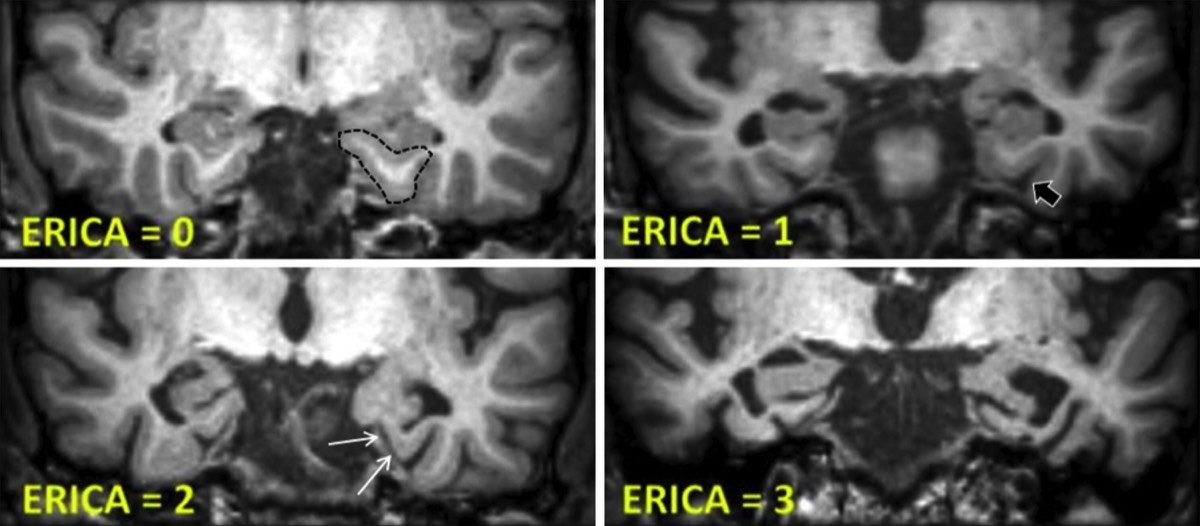
AD病の初期では嗅内野(嗅内皮質)の萎縮が重要なため、entorhinal cortical atrophy (ERICA)が開発されている。
ERICAは、脳幹軸に平行な面で海馬を通り、橋前部レベルで冠状のT1強調画像を用いて行われる視覚的なスコアである。
0:内嗅皮質と海馬傍回の体積が正常
1: 側副溝の拡大を伴う軽度の萎縮(黒矢印)
2: 中等度の萎縮と嗅内皮質が小脳テントから離れている(”tentorial cleft sign"、白矢印)
3: 海馬傍回の顕著な萎縮と嗅内皮質と小脳テントリウムの間の広い隙間
Voxel-based morphometry (VBM)
VBMは脳の萎縮や肥大など体積の変化を客観的に評価しようとするアルゴリズムである。本来はグループ間の比較を行う目的で開発された。VSRAD(Voxel-based Specific Regional analysis system for Alzheimer's Disease)は、側頭葉内側の構造物( 嗅内野、海馬)に関心領域を設置し、相対的な萎縮をz値で示すソフトで、VBMを個々の症例の診断のサポートに用いる画期的なソフトである。視覚的なスコアー化は検者の個体差がどうしても出てくるのに対し、VSRADを用いれば検者の経験不足を補うことができる。
一方で、ADの診断を側頭葉内側の構造物の萎縮のみに頼ることには限界がある事はよく知られている。ADの症例群の中で、海馬の萎縮が軽微な症例(hippocampal sparing type; 注1)は、AD全体の15~20%存在し、特に若年発症に多いと言われている。このような事から、VBMによるADの画像診断アルゴリズムとして、脳全体に関心領域を置いたBAAD(Brain Anatomical Analysis using Diffeomorphic deformation)が開発された。数百にも及ぶ関心領域のz値を提示されても、ヒトの能力では判断が難しくなるため、BAADは人工知能を搭載してAlzheimer’s disease score(ADS)として0〜1の範囲で提示する。この結果、ADSが0.5を超える場合はADの可能性が高いと判断できるようになった。BAAD-AIは放射線科医の診断能力を上回る結果が報告されている。
注1: Ferreiraらの定義(Sci Rep, 2017; 28417965)では、MTAスコアが0で頭頂葉および/または前頭葉の萎縮のあるもの。
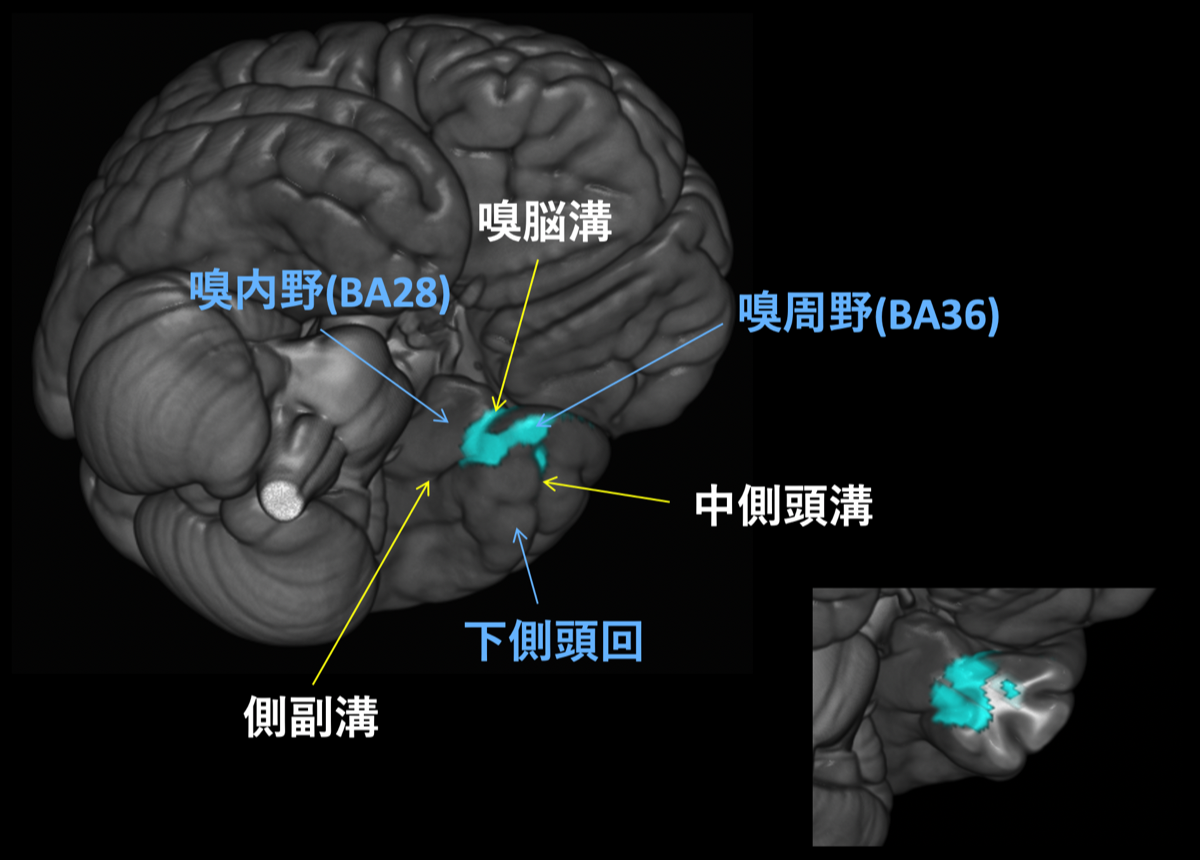
進行性MCIの萎縮部位
MCIからADに進行する際に最も共通して萎縮が認められる領域をVBMで解析した結果、側副溝の前方側周囲の嗅内野と嗅周野、中側頭溝の一部に有意な萎縮を認めた。
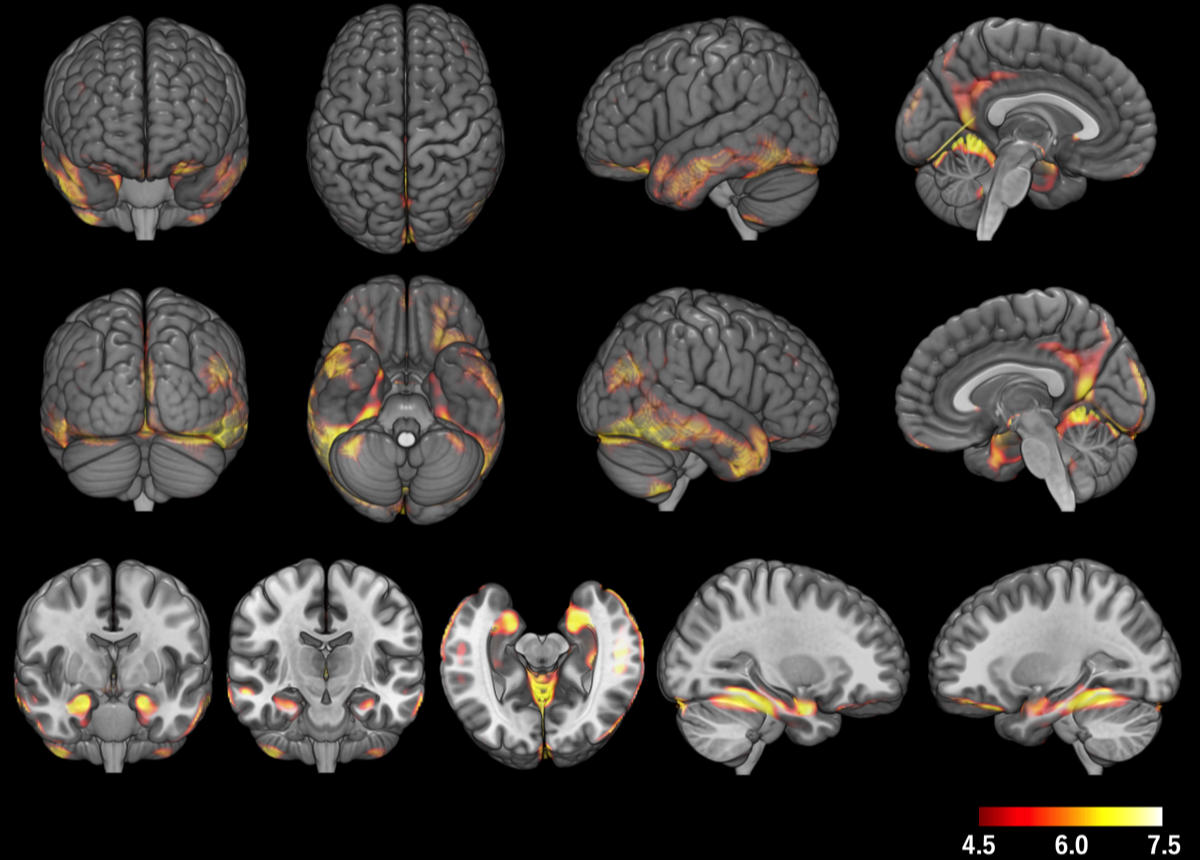
海馬の萎縮が目立たないタイプ
海馬の萎縮はなく、扁桃体、側頭葉外側と底面、頭頂葉、後部帯状回、前頭葉底面の萎縮が見られる。
なお、FWE peak<0.5のしきい値はT=4.5、reselは929 voxelsであった。
アルツハイマー病における萎縮パターンの4種類
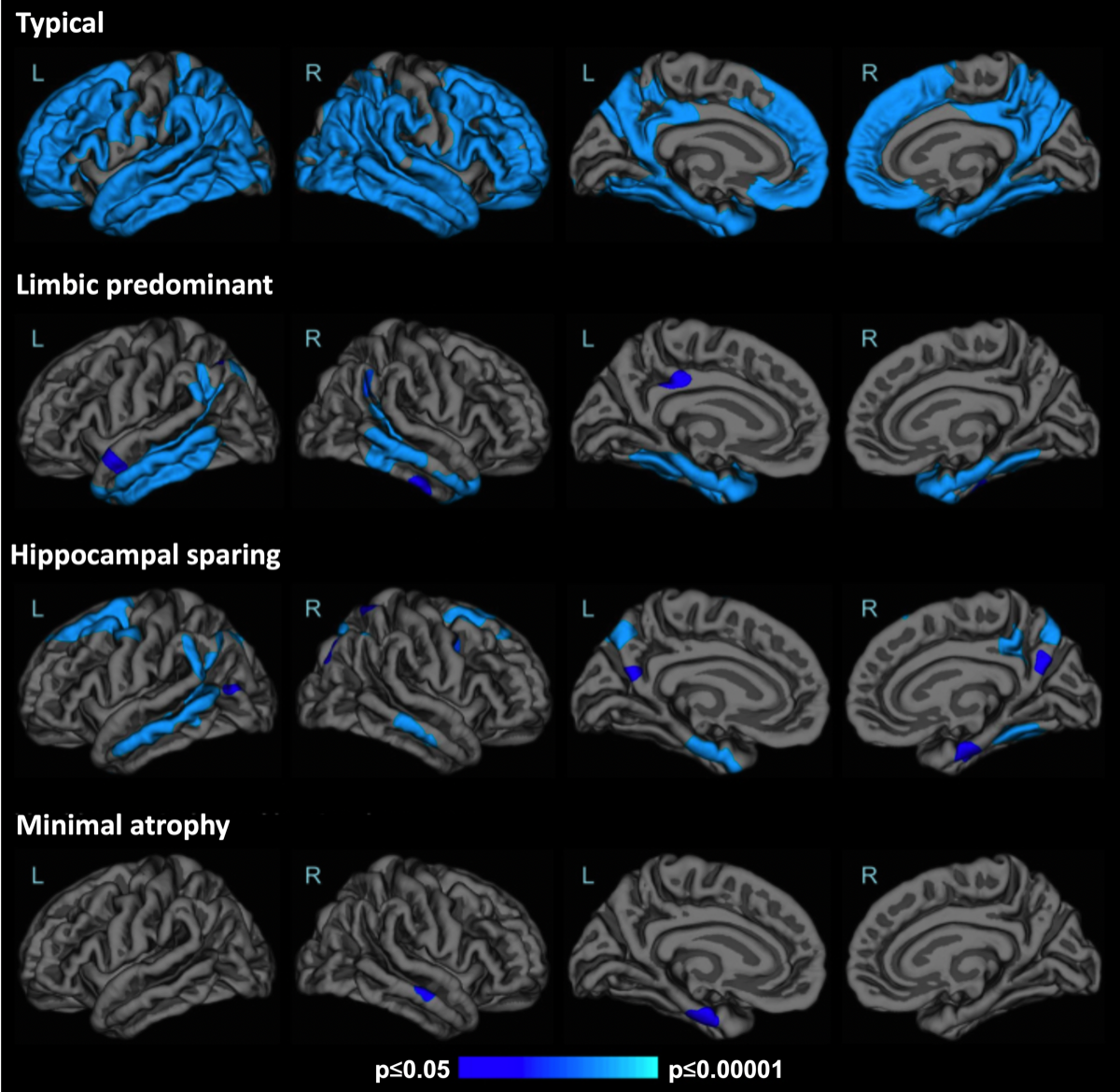
(Ferreira D. et al., Sci Rep, 2017) {28417965}
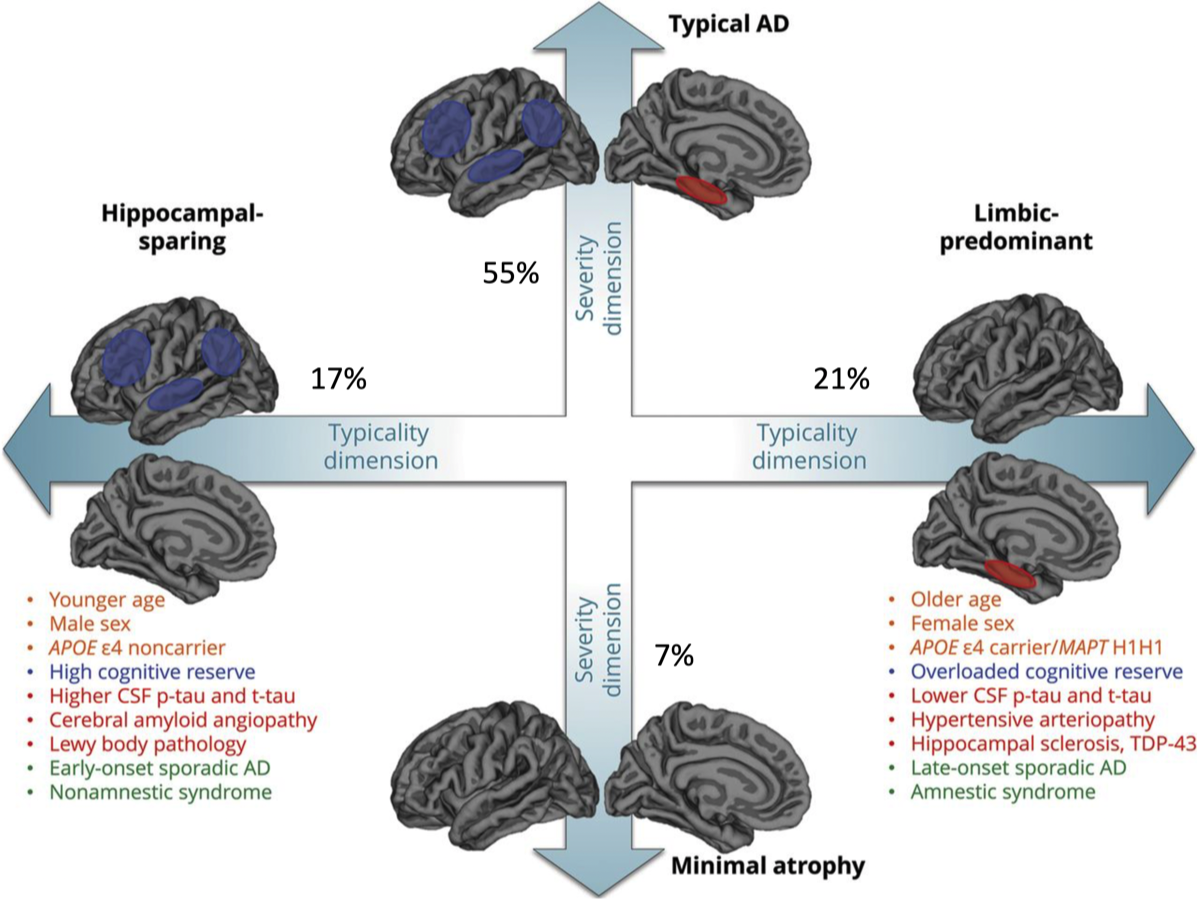
(Ferreira D. et al., Neurology, 2020) {32047067}
Single photon emission tomography (SPECT)による脳血流検査
AD患者を対象としたグルコースPET(FDG-PET)および脳血流SPECT研究により、ADの初期で後部帯状皮質および楔前部におけるグルコース代謝の低下および脳血流低下が出現し、続いて後部視床-頭頂皮質の減少が見られるようになる。これらは両側性でしばしば非対称であり、前頭葉の低下は病期のステージが進行すると認められるようになる。診断精度はFDG-PETで80%前後、脳血流SPECTで70%前後であり、検査にかかるコストや放射線被曝を考えると検査の適応には配慮が必要と思われる。前頭葉の機能低下は中等度のADでも認められるが、この場合であっても機能低下の中心は側頭葉から頭頂葉、後部帯状回であるため、臨床的に前頭葉の機能低下が目立つ症例で、前頭側頭葉性認知症の確認の目的で使う事には意味があると思われる。やや古い報告であるが、99mTc-HMPAO-SPECTのADと血管性認知症の鑑別の感度は71.3%、特異度は75.9%であった。また、ADとFTDの鑑別における感度は71.5%、特異度は78.2%であった。(Dougall NJ. et al., Am J Geriatr Psychiatry) {15545324}.
脳血流SPECTの解析ソフトとしてthree-dimensional stereotactic surface projections(3D-SSP)、easy Z score imaging system (eZIS)、 Z-score summation analysis method (ZSAM)が知られている。石井らによるZSAMの報告では、mini-mental state examinationスコアが24点以上および20~23点の患者において、ADとレビー小体病(DLB)、非ADとDLBの鑑別感度は、それぞれ78.0、88.4 %であり、特異度はそれぞれ44.4、60.0 %であった。また、診断精度はそれぞれ、72.9%、84.2%であった(Ishii K., et al., Jon J Radiol, 2014){24838777}。
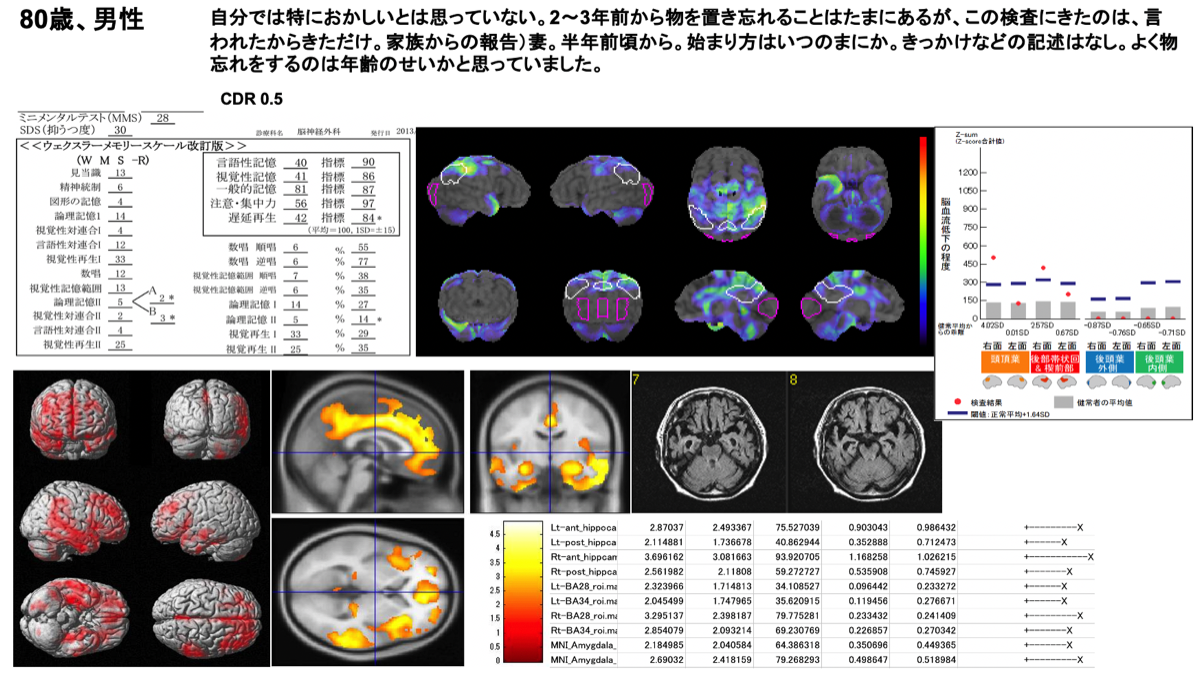
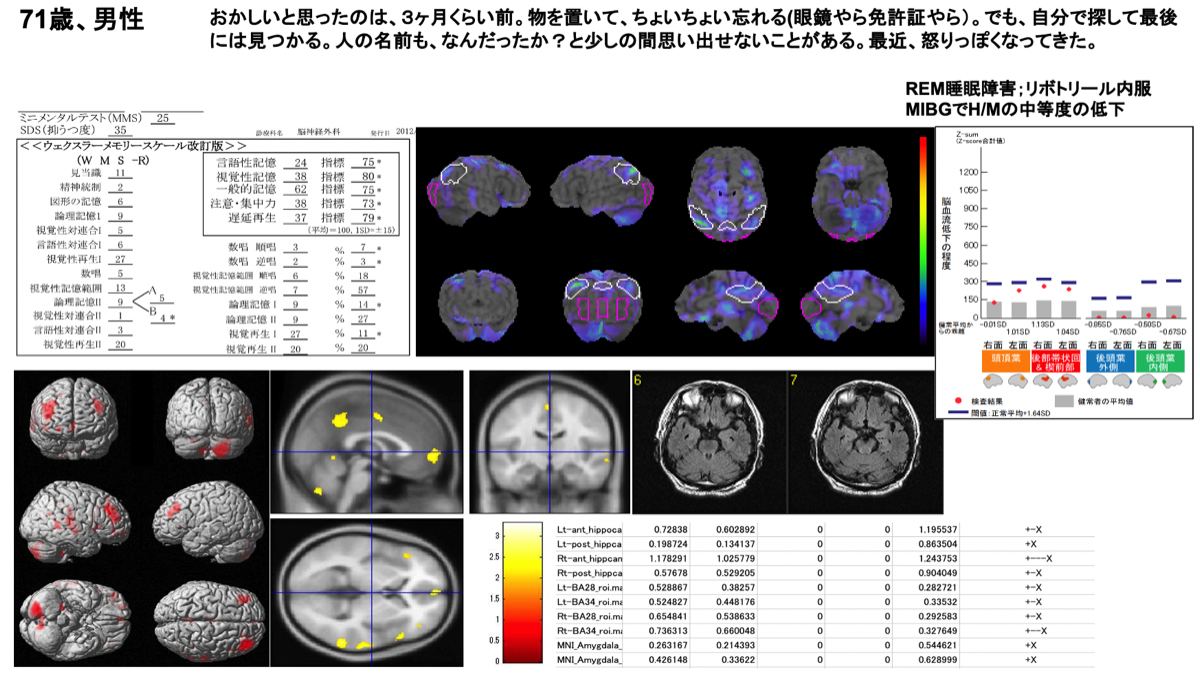
DLBの画像検査は、後述のDAT SPECTが保険適応を取得しており、脳血流SPECTよりも病態特異性は高いと思われる。保険適応はないが、4Rタウオパチーとの鑑別にはMIBGが参考になる。ZSAMを実施した2例の結果を示す。脳血流の低下とVBMで示される脳萎縮の領域に関連があることに留意されたい。
AD初期(CDR=0.5)
DLB(CDR=0.5)
アルツハイマー病の病理診断基準
AD関連病理の神経病理学的評価には、神経斑(CERAD)と神経原線維病理(Braak and Braak)の評価に基づく2つの主要なアプローチがあった。これらの基準を参考に、1997年に米国国立老化研究所(National Institute on Aging; NIA)とレーガン研究所のワーキンググループが統合した基準を勧告した。
米国国立老化研究所(NIA)とアルツハイマー病協会(Alzheimer's Association; AA)が提出したADの神経病理学的評価のための新しいガイドラインでは、ADの神経病理学的変化は、認知機能障害がなくても起こる可能性があることを認識し、アミロイドβ沈着の病理組織学的評価(A)、神経原線維変化のステージング(B)、神経斑のスコアリング(C)の「ABC」スコアに従って評価することが推奨されている。
なお、18F-flutemetamol PETによるプラーク検出の下限は、CERADのsparseとmoderateの間にあるとされている(Ikonomovic MD. et al., Alzheimers Res Eher, 2018) {29935545}.
NIA-AAのABC score
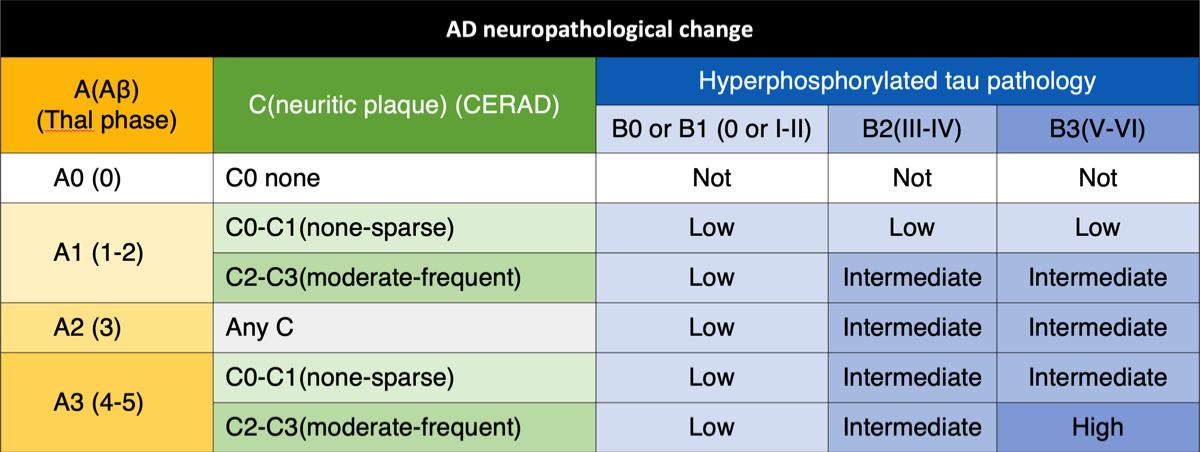
Amyloid plaques:アミロイドβの細胞外沈着物で、AD患者の大脳皮質に豊富に存在する。アミロイド斑は、その形態とThioflavin-SやCongo Redでの染色に基づいて、diffuse plaqueとdense-core plaqueに分類される。
Dense-core plaque:Thioflavin-SやCongo Redで染色されたコンパクトなコアを持つ線維状のアミロイド沈着物。典型的にはdystrophic neurite(neuritic plaques)、反応性アストロサイトおよび活性化ミクログリア細胞に囲まれており、シナプスの喪失と関連している。Neuritic plaquesの存在は一般的に認知障害の存在と関連している。
Diffuse plaques:Thioflavin-SやCongo Red陰性で、不明確な輪郭を持つ非晶質アミロイド沈着物。これは通常、非神経症性であり、グリア反応やシナプスの喪失とは関連していない。このプラーク型は、正常の高齢者の脳に比較的よく見られる所見であるため、ADの病理診断には考慮されない。
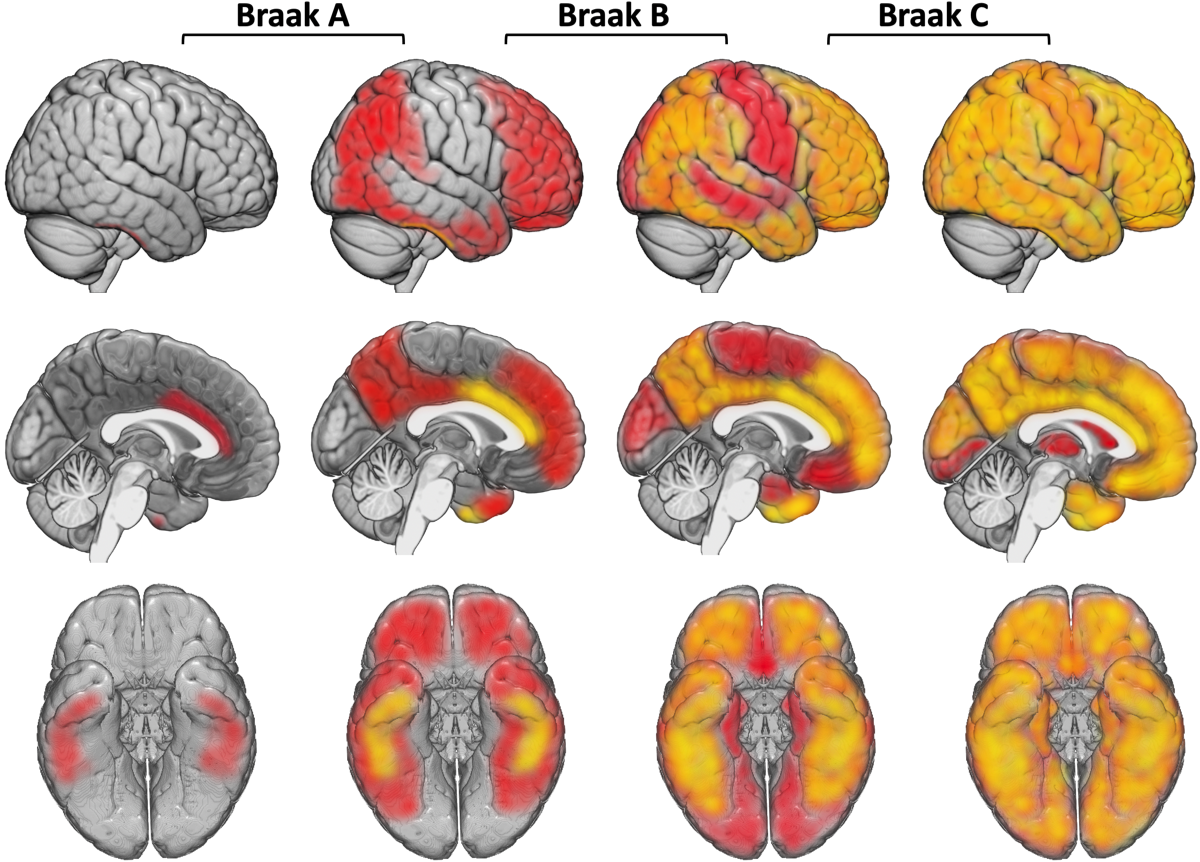
アミロイドβの細胞外沈着である老人斑の広がりはNFTとは異なる経過をたどる。また老人斑は健常者でも加齢とともに認められる。Thalのphase分類では、老人斑は連合野に始まり、前頭葉、側頭葉、後頭葉の底面の皮質3層を中心に出現し始める。この段階では海馬には老人斑は出現していない。次いで、海馬領域を含めた辺縁系、基底核と視床、一次知覚・運動野に及び、最終的には視床下部、脳幹、小脳に広がる。なお、新皮質での老人斑の広がりはBraakのステージ分類としてA、B、C分類が知られているが、最近ではアミロイドPETを用いた分類が注目されている。PET研究では、Aβ蓄積は、楔前部、後部帯状回、島皮質、内側および外側眼窩前頭皮質に蓄積し、そこから側頭葉底面を含めた大脳皮質全体に広がる。

アルツハイマー病の病態
アルツハイマー病(AD)においてはアミロイドβ沈着による多数の老人斑以外に、タウ病変による神経原線維変化という2つの病変が同時に見られる。実際に神経細胞の変性に関わっているのは神経原線維変化と考えられている。家族性アルツハイマー病の遺伝子異常のほとんどがアミロイドβの生成に関係している事から、アミロイドβはADの発症に関係しているとされている。アミロイドβの沈着が何らかの理由で神経細胞に神経原線維変化を引き起こす、というのがアミロイドカスケード仮説。カスケードとは連なった小さな滝を意味しているが、アミロイドβの蓄積から神経原線維変化を引き起こすまでには複数の因子(小さな滝)が関係している可能性がある。
アミロイドβが脳に蓄積しているからといって必ずしもADを発症するとは限らない。家族性アルツハイマー病の場合は100%発症するので矛盾しているように思えるが、アミロイドβと神経細胞の変性(タウ病変)の直接的な因果関係は証明されていない。例えば家族性アルツハイマー病の1つを再現したAPP(amyloid precursor protein)の遺伝子改変マウス(Tg2576)では、多数の老人斑が出現するがタウ病変は再現できない。これには様々な説明があるが、少なくとも仮説の域を出ていない。
興味深いことにBraakらの報告では{22002422}、アミロイドβの蓄積前からタウ病変は始まっており、アミロイドカスケード仮説における、上流にアミロイドβ、下流にタウ病変という関係は絶対的なものではない可能性がある。老人斑は大脳の連合野から始まるのに対し{23137949}{12084879}、神経原線維変化は側頭葉内側から始まることも、双方の病態が別々に起きていることを示唆している。18F−AV45 PETによる研究では{29046362}、側頭葉底面と前部帯状回へのアミロイドβの沈着が早期に認められる。側頭葉底面はタウ病変が新皮質にはじめに進展する領域であり、この場所でタウ病変がアミロイドβに遭遇することがアルツハイマー病の起点となっている可能性がある。すなわち、上流と下流の関係ではなく、並行して流れている川が合流するイメージとなる。
アミロイド前駆物質(APP; amyloid precursor protein)
アミロイドとは不溶性になったタンパク質が蓄積して線維状の構造を形成したもので、その構造物はアミロイド線維(amyloid fibril)と呼ばれる。ADに限らず、クロイツフェルトヤコブ病、透析アミロイドーシスなど細胞外に蓄積するもの、パーキンソン病やハンチントン病のように細胞内に蓄積するものがある。ADではアミロイドのうちアミロイドβタンパクがクロスβ構造をとり細胞外に蓄積している。
アミロイドβはアミロイド前駆蛋白(APP)から作られるが、APPが何の目的で作られているのかは明らかにされていない。ヒトではAPPと類似したタンパクにAPLP-1(APP-like protein-1)とAPLP-2(APP-like protein-2)が知られており、APPと共に生体での機能を相互に補っていると考えらている{25710536}。APPやAPLPは神経細胞内での軸索移動やシナプス形成に関わっていることや{17047360}、胎生期の神経前駆細胞の移動(migration)に関係すると言われている。
APPからアミロイドβ(Aβ)の生成機構
APPは図のように細胞膜を1回貫通するタンパク質で膜上を自由に移動している{20107219}。(1)APPが細胞膜(小胞膜)上でα-セクレターゼに遭遇するとsAPPαとして切り出され、その後γ-セクレターゼで分解されてP3となり、アミロイドβは作られない。(2) APPがβ-セクレターゼ(BACE-1)で切断されるとsAPPβが形成され、その後γ-セクレターゼによってアミロイドβが作られることになる。この際にAICDと呼ばれる細胞内ドメイン(APP intracellular domain)が作られる。
*ラフト(筏)とは、細胞膜上を自由に漂流するスフィンゴミエリンやコレステロールを多く含んだ領域で、レセプターなどのタンパク質が多く存在する。細胞膜上での信号伝達にはこのラフトが重要な役割をもっている。APPのβ-セクレターゼの切断は、主にこのラフトの領域で行われている可能性がある。ApoE4がラフト領域を拡大し、アミロイドβの産生を増加させる可能性はあるが、不明なことが多い。少なくとも、脳のアミロイドβ種の蓄積はApoEアイソフォームに依存する(ε4>ε3>ε2)というモデルが支持されている。
細胞膜(小胞体膜)におけるアミロイド先駆物質(APP)の代謝
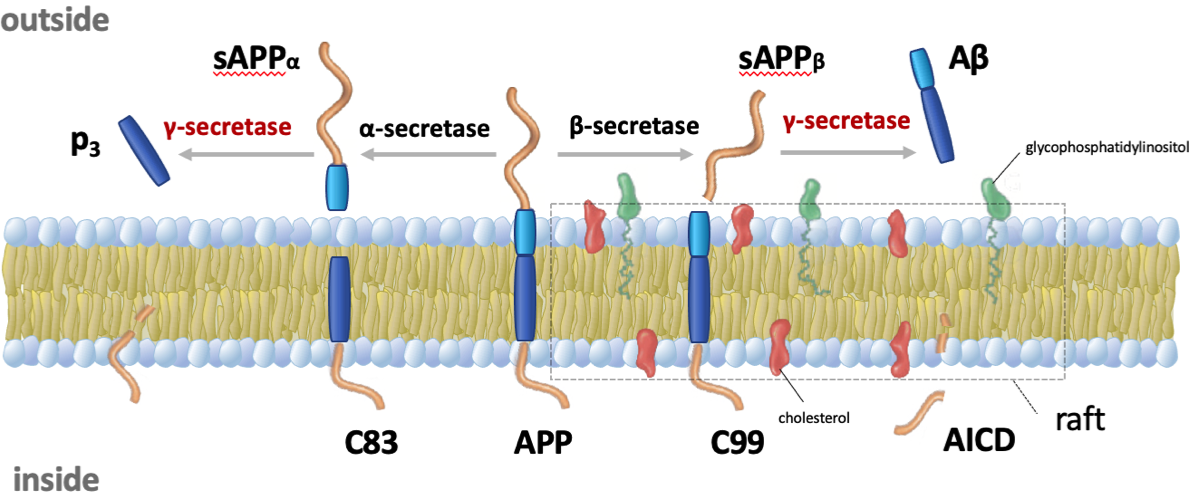
APPがはじめにα-セクレターゼで分解されると、アミロイドβは産生されない。ラフトでβ-セクレターゼにより分解されると、次にγ-セクレターゼで分解されてアミロイドβが作られる。この際に細胞内ドメイン(AICD)が切り出される。Notchシグナルと同じように、AICDは遺伝子発現に影響すると考えられている。γ-セクレターゼを修飾する薬剤は、今のところ、アルツハイマーに有効性が証明されていない。
最近の研究では、これらの反応が全て細胞表面の膜(plasma membrane)で起きているのではない事がわかっている。APPが細胞内で合成され細胞表面の膜まで輸送されるとα-セクレターゼによって切断される。一方、β-セクレターゼの作用のほとんどは細胞内小胞の膜上で起きると考えられている。この反応は合成されたAPPが細胞表面に輸送されるまでに起きている可能性と、一旦、APPが細胞表面まで運ばれた後にエンドサイトーシスによって細胞内に取り込まれてから起きている可能性がある。いずれにしても、アミロイドβとAICDは細胞内で作られ、1)細胞外に排出、2)細胞内で分解、3)核内に取り込まれ遺伝子発現に作用する可能性が指摘されている。γ-セクレターゼはNotch蛋白を切断して、細胞内ドメイン(NICD)を細胞内に放出させる機能も持っている。NICDは核内に入り、複数の標的遺伝子の発現を誘導して細胞の分化を促すことから(Notch signaling)、AICDも同じように遺伝子発現に関与すると考える研究者がいる。最近の研究によると、AICD単独では機能を発揮することはないが、AICD-Fe65-Tip60というAFT complexを核内で形成して何らかの作用を引き起こすのではないかと推測されている。また、アミロイドβも核内に入り遺伝子の発現に影響を与えているとする報告もある{26296890}。
家族性アルツハイマー病は常染色体性優勢遺伝を示し、その割合は全アルツハイマー病の1%程度。こうした家系における遺伝子の異常は、第21番染色体のAPP遺伝子、第14染色体のプレセニリン-1、第1染色体のプレセニリン-2遺伝子であることがわかっている。1つ目のAPP遺伝子の異常はAPPのアミノ酸配列に異常をきたすもので、アルツハイマー病以外にも脳アミロイドアンギオパチー(CAA)を引き起こす。変異したAPPはβ-セクレターゼへの親和性が高く、アミロイドβが多く産生され、かつ、アミロイドβ1-42(通常はアミロイドβ1-40)の割合が高くなっている。また、21トリソミーとして知られるダウン症がアルツハイマー病を発症しやすいのもAPPの産生過剰によるアミロイドβの増加が原因と推測されているが、本当のところは解明されていない。他の遺伝子異常はプレセニリン(γ-セクレターゼの構成蛋白)に関わるもので、いずれもγ-セクレターゼの機能障害が関係している。
γ-セクレターゼの障害とアルツハイマー病
γ-セクレターゼはプレセニリン-1またはプレセニリン-2、PEN-2、APH-1、ニカストリンの4つのサブユニットで構成されているプロテアーゼ(蛋白分解酵素)。最近になりプレセニリンは細胞膜を9回貫通する蛋白であることが判明した{23254937}。プレセニリンはγ-セクレターゼの活性中心であり、そのアミノ酸配列の異常はアルツハイマー病を引き起こす。γ-セクレターゼはAPP、APLP以外に細胞の分化に関係するNotchタンパクも基質としているが、Notchシグナル伝達経路の障害は、様々な発生異常や癌、CADASILなどに関係してる。
図Aは家族性アルツハイマー病のプレセニリン-1におけるアミノ酸配列のミスセンス部位(通常とは異なるアミノ酸に変異)を赤で示しているが、膜貫通部付近での異常が多いことがわかる。プレセニリンの異常はγ-セクレターゼの機能障害を引きおこしアミロイドβの産生はむしろ減少するはずなのに、なぜアルツハイマー病を誘発するのか。
グラフ(図B){17197420}はγ-セクレターゼ阻害剤を投与した際のアミロイドβの生成を模式的に示したもの。阻害剤の投与によりAβ1-40の産生は減少するが、蓄積しやすいAβ1-42の産生は増加している。プレセニリンに障害があるとγ-セクレターゼによるsAPPβの切断位置がズレて、Aβ1-42やAβ1-43が多く産生される。Aβ1-42における増加した2個のアミノ酸はイソロイシンとアラニンで、これらは疎水性のためAβ1-40よりも不溶性で重合しやすく、細胞に対して毒性を持つようになると言われている。
可溶性が低いAβ1-42は主に脳実質に老人斑として沈着するが、可溶性の高いAβ1-40は 脳実質の細動脈(時には細静脈)の中膜壁を介してリンパ管に流出、glymphatic pathwayを介して排出されている。高血圧などにより、細動脈の中膜(平滑筋)の構造が変化するとこの排出機構が低下し、Aβ1-40は中膜に沈着して、脳アミロイドアンギオパチー(CAA)や微小出血を引き起こす。Glymphatic pathwayを介したクリアランス以外に、細胞内のアミロイドβは、ユビキチン-プロテアソーム経路で、細胞外ではネプリライシンを介した経路で、それぞれプロテアソームやアミロイドβ分解酵素(ADE)によって分解される。
軽度認知障害(MCI)でAβが陽性の場合は、prodromal ADあるいはMCI due to ADと呼ばれ、アルツハイマー病の発症前=治療のターゲットと考えられている。健常者でも高齢になるとアミロイドβの陽性率は高くなり、80歳を超えると65%が陽性になると報告されている(5)。HDLやVLDLなどを構成するアポリポ蛋白の3つの対立遺伝子(ε2、ε3、ε4)のうち、ε4はAβの沈着を促進すると言われている。また糖尿病もアミロイドβの沈着と関係していると言われている。一方、ε2はタウオパチーと関連しており、ε2/ε2遺伝子型が進行性核上性麻痺(PSP)の危険因子であることが示されている。さらに、APOE ε2対立遺伝子は一般人口よりも嗜銀顆粒性認知症(AGD)患者に多く見られる。
老人斑(アミロイドβの沈着)はアルツハイマー病だけに見られるのではない。例えばレビー小体病の30%以上にアルツハイマー病と同じ病理所見が認められる。認知症でない健康者にも老人斑は認められ、非認知症とアルツハイマー病との間で老人斑の出現の程度や分布に明確な境界はない。
アミロイドPETは、脳のAβの沈着を反映し、前頭側頭型認知症(FTD)とそれ以外の変性型認知症との鑑別に有用とされてきたが{18690556}、病理でFTDと診断された症例の1/3にアミロイドβの沈着が認められ、以前考えられていたように明確に鑑別できるものではない。アミロイドPETが陽性であっても、病理でFTD-tauの症例が存在する{28653036}。定義上、アミロイドβの沈着のないアルツハイマー病は存在しないが、アミロイドβの沈着があってもその程度が軽い場合には、アルツハイマー病以外の認知症の可能性は否定できない。
図A: プレセニリン-1の遺伝子異常
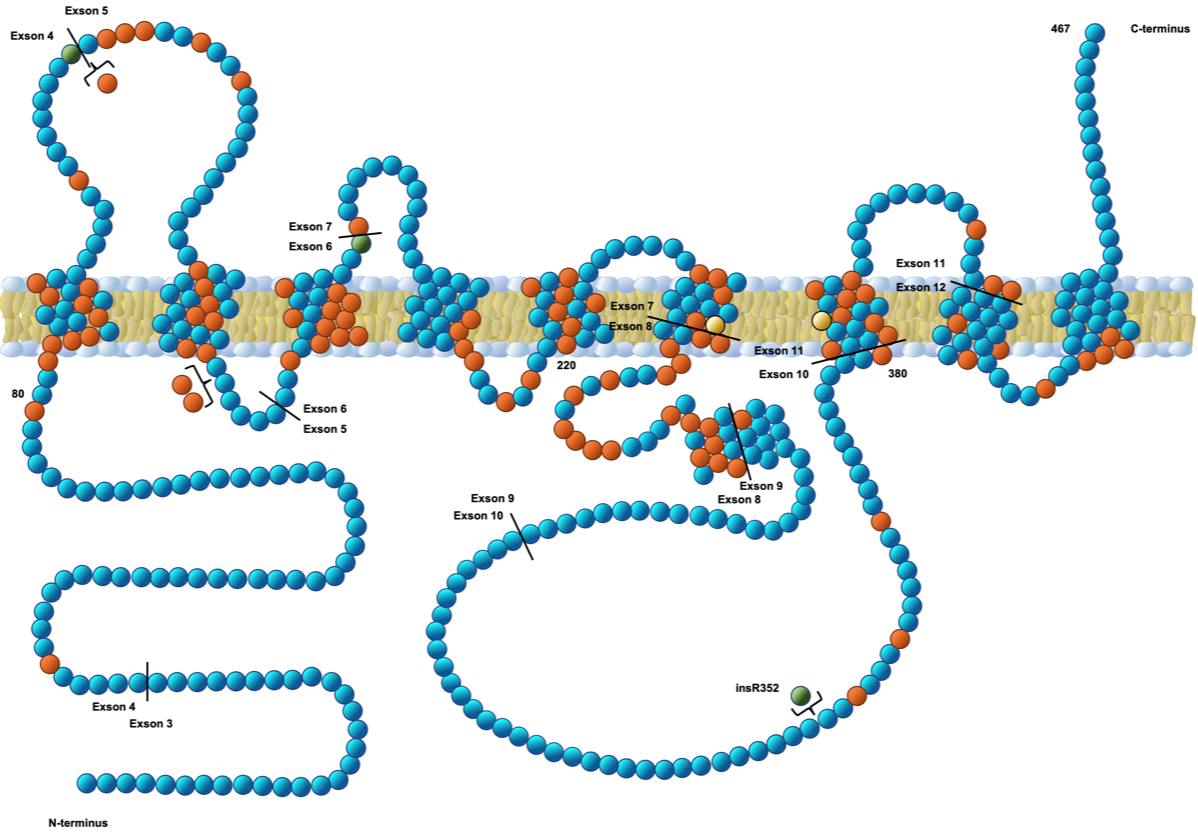
プレセニリン-1はγ-セクレターゼを構成する4つのサブユニットの1つであるが、そのアミノ酸配列の異常は家族性アルツハイマー病を引き起こす。赤は家族性アルツハイマー病で異常が見つかっている部位で、膜貫通部位に集中している。緑は(L113P, G183V, and insR352)の変化はPick病を引き起こし、アミロイドβの沈着は認められない。黄色(アスパラギン酸)はγ-セクレターゼの活性に重要な部位を示している。
図B: γセクレターゼ阻害とAβの産生
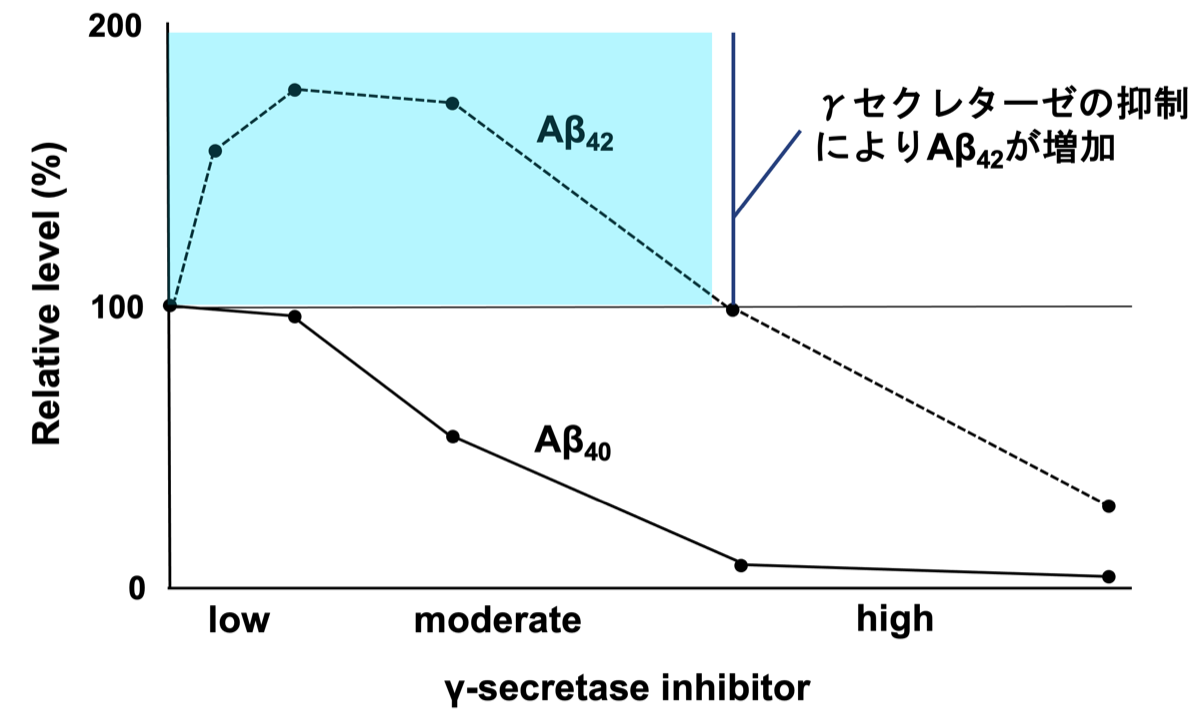
セクレターゼの作用を阻害すると、細胞に有害なAβ42の産生が増加してしまう。
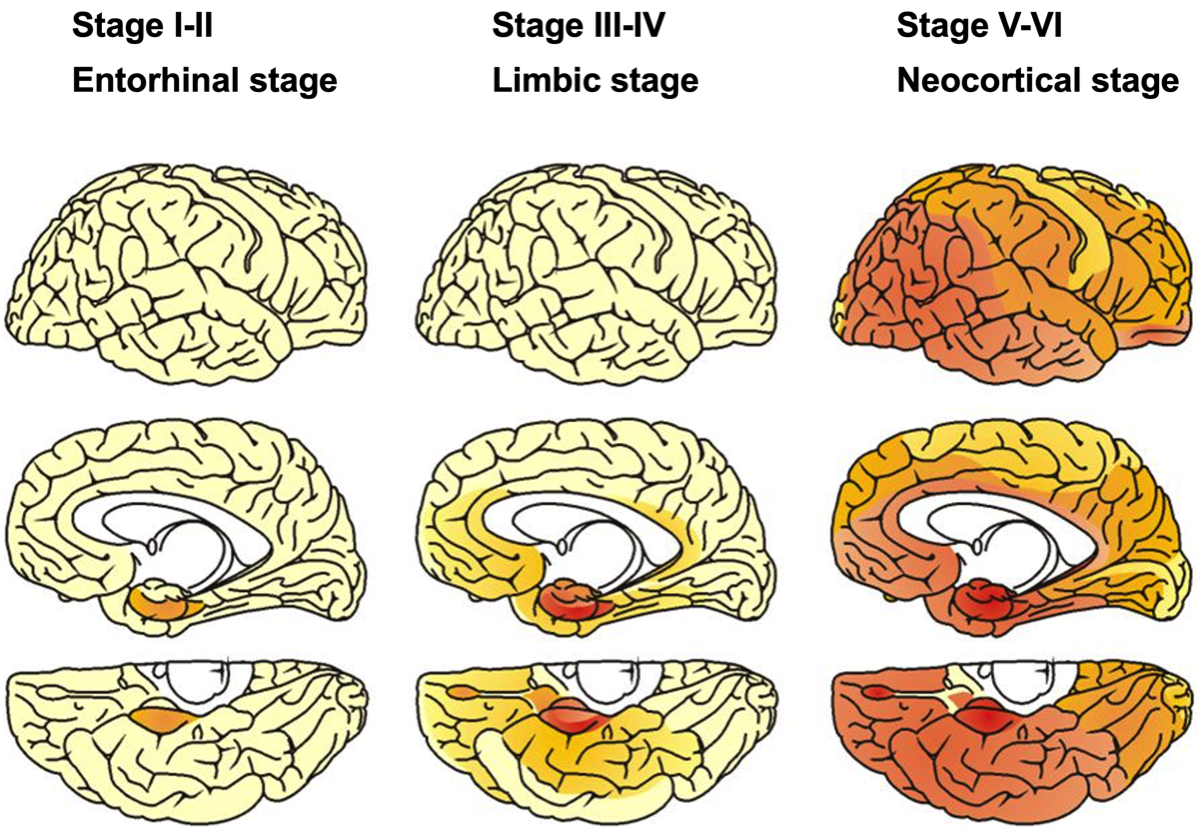
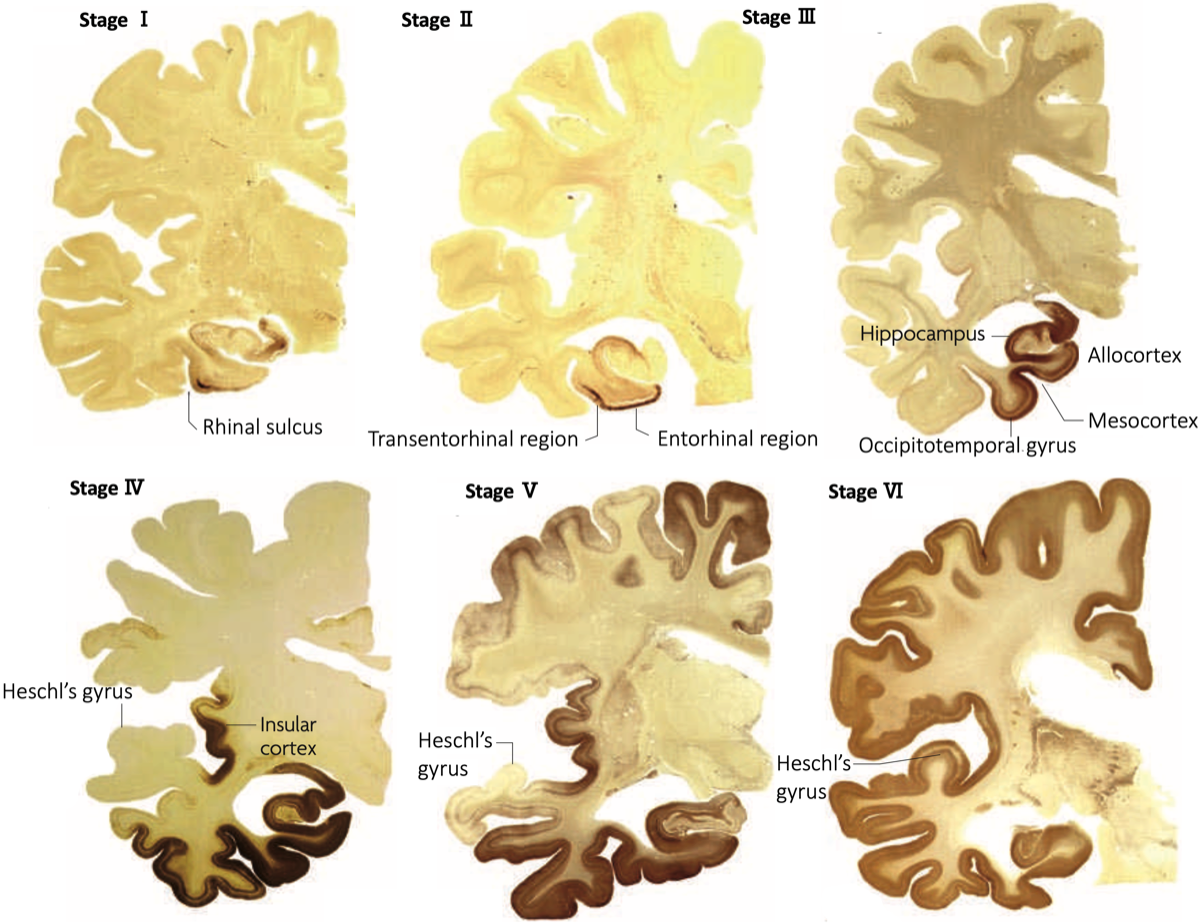
(Braak H., rt al., Acta Neuropathol, 2006) {16906426}
(Braak H., rt al., Acta Neuropathol, 2006) {16906426}
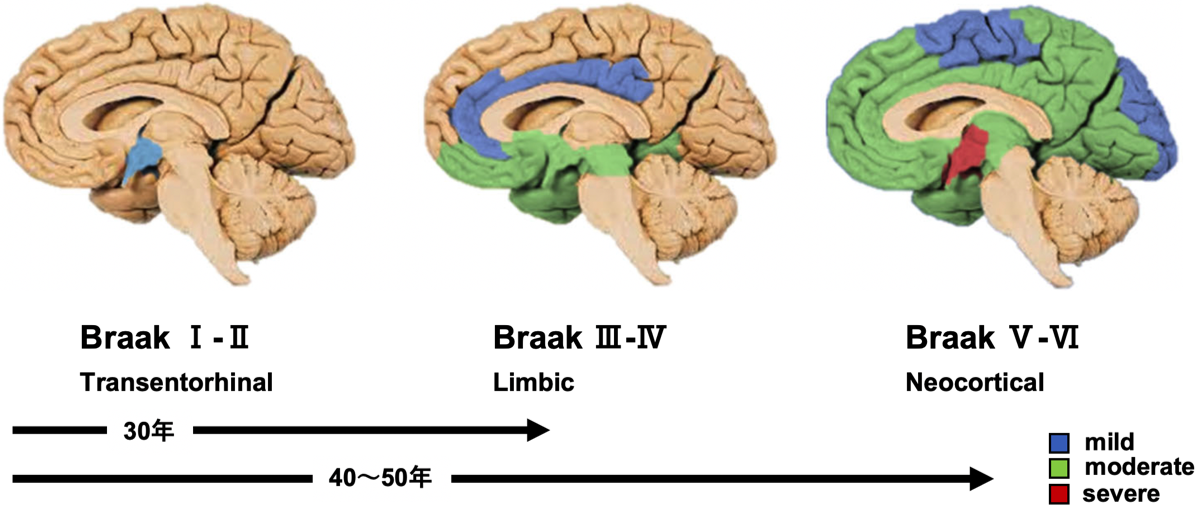
Stage I:嗅脳溝(rhinal sulcus, 側副溝の続きで前方に位置する)の経嗅内野(transentorhinal region)のPre-α細胞(II層) に出現。ごく少数のNFTは海馬CA1、基底前脳の大細胞核や視床の前背側核にも散在。
Stage II:内側の嗅内野(entorhinal region)に進展。海馬体ではCA1〜海馬台移行部に少数出現する。ごく少数の弧発性のNFTが等皮質の連合野に出現してもよい。
Stage III:経嗅内野と嗅内野のPre-α(II層)は高度に冒され、経嗅内野Pre-αにはゴーストタングルがみられることがある。Pre-β(Ⅲ層)やPri-α(Ⅴ層)細胞にも少数のNFTが認められるようになる。海馬台の錘体細胞には尖端樹状突起までのびるNFTが出現するようになる。CA2〜CA4では少数の細胞を除いて変化をまぬがれる。新皮質では症例によっては前頭、側頭、後頭連合野の底部のIII, V層に少数のNFT、neuropil thread (NT; 樹状突起や軸索にリン酸化タウが蓄積したもの)が散在する。多くの例で基底前脳大細胞核、視床前背側核、扁桃体に軽度の変化がある。弧発性のNFTが視床結合核, 視床下部の乳頭隆起核にみられることがある。
Stage IV:経嗅内野、嗅内野Pre-αは非常に高度に冒され、多数のゴーストタングルが現れる。CA4の歯状回近傍に多極性細胞の星形NFTが現れるようになる。新皮質はごく軽度の変化にとどまり、一次運動・知覚野に変化はない。扁桃体ではNFT、NTはおもに腹外側核に出現する。前障の基底部は軽度に冒される。被殻と側坐核の基底部にNFTがみられることがある。視床前背側核はNFTとNTで密に満たされ、結合核や視床下部の乳頭隆起核もより強く冒される。
Stage V:海馬はすべての部位が冒される。新皮質が強く冒されるが、所見がまだ軽い段階では変化は側頭〜後頭葉の内側基底部および底面、次いで、島と眼窩皮質の前基底部が冒される。より高度になると上側頭回を除く領域にNFTとNTが多量に出現する。すべての連合野が冒されるが一次知覚野は抵抗性があり、一次運動野は新皮質のなかでは最も冒されにくい。皮質下神経核の変化はstage Vでより明瞭になる。扁桃体に接する前障の基底部は恒常的に冒される。視床の前背側核には多数のゴーストタングルが現れ、前腹側核にも最初の変化がみられるようになる。少数のNFTとNTが視床下部の外側隆起核や黒質の緻密層に観察される。
Stage VI:しばしばゴーストタングルは除去されてグリアに置き換わっている。歯状回の顆粒神経細胞の無数のNFTはstage Vとの鑑別点である。すべての新皮質連合野は非常に高度に冒される。一次感覚野ではV層にNFTは少ないが密なNTが境界明瞭な帯を形成し、stage Vとの鑑別点となる。これに対して一次運動野のV層に所見はみられない。皮質下神経核は非常に高度に冒される。視床の前腹側核を被う嗜銀性の神経突起網は網様核にまで延びる。前背側核は特に高度に冒される。視床下部の外側隆起核にNFTがみられることがある。Stage VIは錐体外路系が冒される点が特徴的である。線条体のほとんどの大型細胞とかなりの数の中型細胞がNFTを示す。さらに、長く延びた樹状突起をもつNFTが黒質の多くのメラニン含有細胞に認められる。
タウ蛋白は神経細胞の軸索にある微小管に付着してこれを強化している。その遺伝子は第17染色体(17q21)のMAPT(microtubule-associated protein tau)に存在する。16個のエクソンのうち、E2、E3、E10の3つのエクソンにおけるm-RNAのスプライシングの違いによって6種類のタウ蛋白が作られる。微小管に結合する部位を微小管結合領域(microtubule-binding domain; MBD)と呼ぶが、この数が3つのものは3R(リピート)タウ、E10が挿入されて4つになると4Rタウと呼ばれ、4Rタウの方が微小管への結合力は強いことになる。興味深いことに、ヒトの胎生期には3Rタウのみが発現しており、大人になるにつれて4Rタウが増えてくる。タウ蛋白の微小管への結合はリン酸化によっても調節を受けており、タウ蛋白がリン酸化を受けると微小管から遊離して樹状突起などに移動する。何らかの理由でタウ蛋白が過剰にリン酸化を受けると、細胞内でタウ蛋白が重合して沈着する。MAPT遺伝子にはH1とH2の2種類のハプロタイプがあり、H1の多い方が認知症になりやすいと言われている{26444794}。
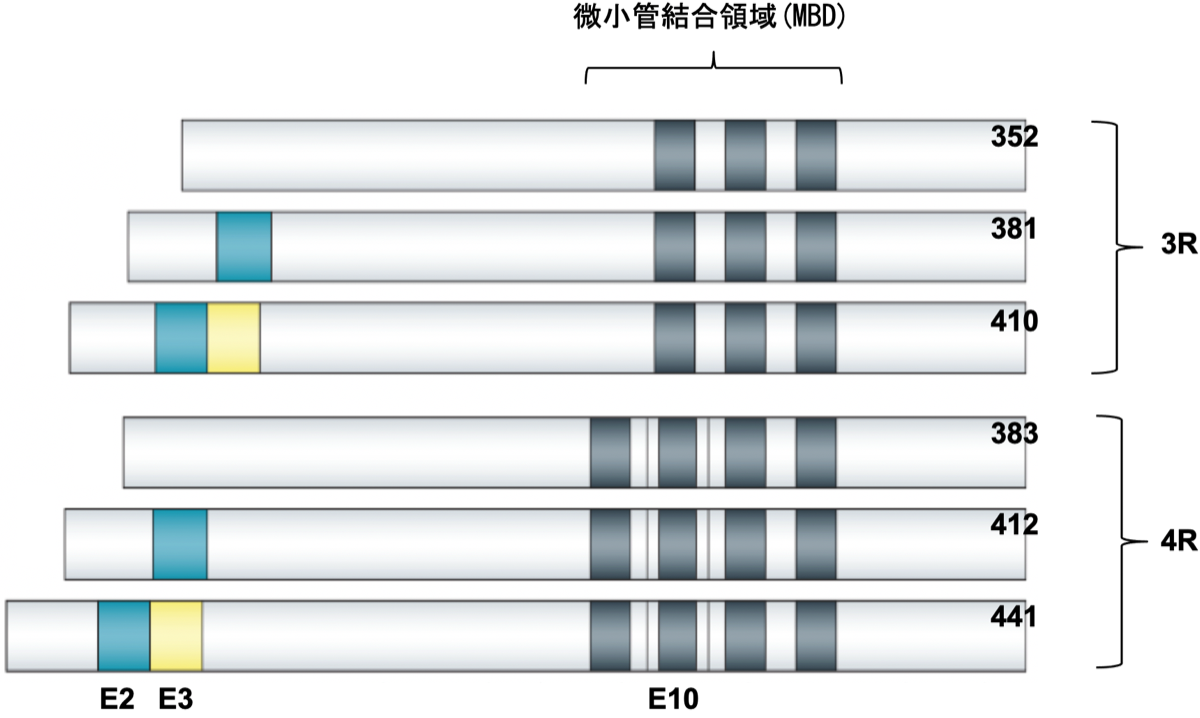
アルツハイマー病では3Rと4Rが1:1の比率で混ざった過剰リン酸化タウが神経細胞内に沈着して神経原線維変化として出現している。この比は正常に発現しているタウと同じ比率なので、通常のままの割合で沈着しているように見える。軽微な脳の外傷が神経原線維変化を引き起こすこと{28742910}、 外傷はアルツハイマー病の危険因子であること(オッズ比3.3){10746604}は以前より知られているが、これはchronic traumatic encephalopathyと呼ばれている。超高齢者に多いprimary age-related tauopathy (PART)は、老人斑のないこと以外はアルツハイマー病のタウ病変と同じで、加齢に伴い神経原線維変化が出てくると思われる。一方、前頭側頭型認知症として有名なピック病では神経細胞内に過剰にリン酸化した3Rタウが凝集してピック小体として認められる。比較的明瞭な境界をもって前頭葉と側頭葉前部(前部連合野)の萎縮が認められのが特徴。進行性核上性麻痺(progressive supranuclear palsy; PSP)や皮質基底核変性症(corticobasal degeneration; CBD)では4Rタウが沈着している。これらの疾患では、アストロサイトやオリゴデンドログリアにも4Rタウの沈着が認められる。嗜銀顆粒性認知症 (argyrophilic grain disease; AGD)も4Rタウオパチーに分類されるが、apical dendrite (尖端樹状突起)に蓄積してneuropilに顆粒状に認められることが特徴。このようなAGDの病理所見は高頻度にPSPやCBDの症例にも認められている。Astrocytic plaqueはCBDに、tufted astrocyteはPSPに特徴的とされているが、これら3疾患には共通点が多い。
タウのリン酸化部位は2つのタイプに分けられる。CDK5, GSK-3, mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK)などのセリン/スレオニンプロリン誘導型キナーゼによって修飾される部位と、Microtubules (MT)親和性制御キナーゼ(MARK)などの非プロリン誘導型キナーゼによって修飾される部位である。タウは微小管に結合して安定させ、細胞質の延長の形成、軸索の輸送、外的障害からの保護に関与している。タウの過剰なリン酸化は、タウの微小管からの遊離を引き起こし、細胞質に蓄積、さらに凝集してNFTを形成する。CDK5によるタウのリン酸化は、タウに対するGSK3βのキナーゼ活性を増加させる。すなわち、CDK5によるSer235および404のリン酸化は、GSK3βが介在するThr231およびSer400および396のリン酸化の増加を促進し、GSK3βによるタウのリン酸化が増加する。ADのような病的過程では、カルシウム依存性プロテアーゼであるm-カルパインやμ-カルパインの活性化により、p35およびp39のようなCDK5アクチベーターが阻害され、別のアクチベーターであるp25およびp29タンパク質がCDK5に作用してCDK5の活性化が長引く。細胞内にカルシウムが流入すると、カルシウム依存性のチオールプロテアーゼであるカルパインが活性化され、CDK5の活性化因子であるp35がp25とp10の2つに分断される。p35/CDK5複合体はp35をリン酸化し、ユビキチン化とそれに続くプロテアソームを介したp35の分解を誘導する自動制御シグナルとなるが、p25はCDK5とより安定した複合体を形成してその活性を高め、タウのリン酸化が長期的に増加して神経衰弱を引き起こす。
この過剰な活性化は、タウの過リン酸化を引き起こし、paired helical filaments (PHF)の形成、それに続くNFTの形成、細胞骨格と細胞内輸送の破壊を引き起こし、神経変性を誘発する。
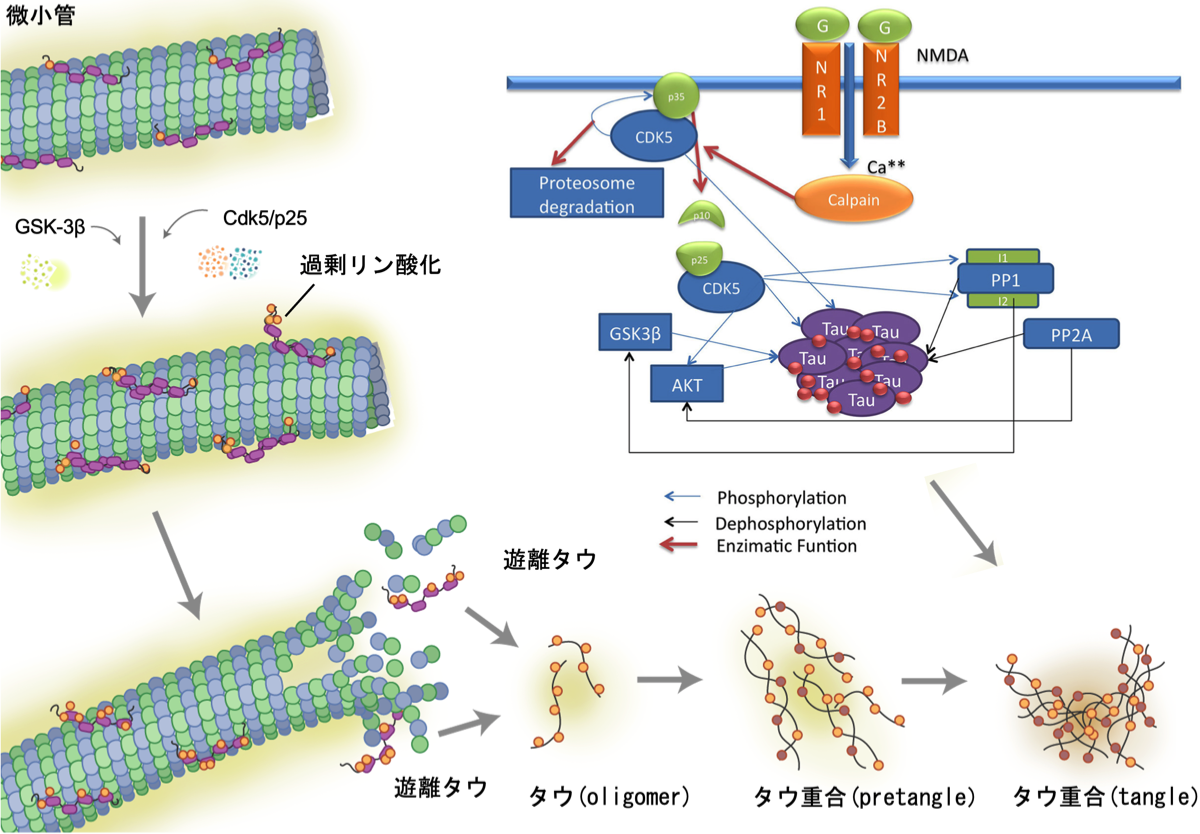
(Querfurth HW. et al., N Enlg J Med, 2010) {20107219}
(Castro-Alvarez JF., et al., Front Aging Neuroscience, 2014) {25225483}
病的なタウ蛋白がプリオン蛋白のように神経細胞から神経細胞に伝播する可能性が指摘されている。仮にそうだとすると、脳内ネットワークは神経変性疾患の進行の鍵を握っているのかもしれない。多くの神経変性疾患の萎縮パターンは、安静時の機能的脳ネットワークに類似していることが示されており、ネットワークの「ハブ」は、脳疾患全体で神経変性の影響を特に受けやすい。タウは、そのままの状態、あるいはエクソソームとして細胞外空間に分泌されることが観察されている。これが別の神経細胞に取り込まれると、病原性のミスフォールドしたタウタンパク質は、「種」として作用しゆっくりと伝播することがタウ病理の脳内拡散の原因であると考えられる。アミロイドβの存在はこのようなタウの伝播を促進すると考えられている(Busche MA, et al., Nat Neurosci, 2020)
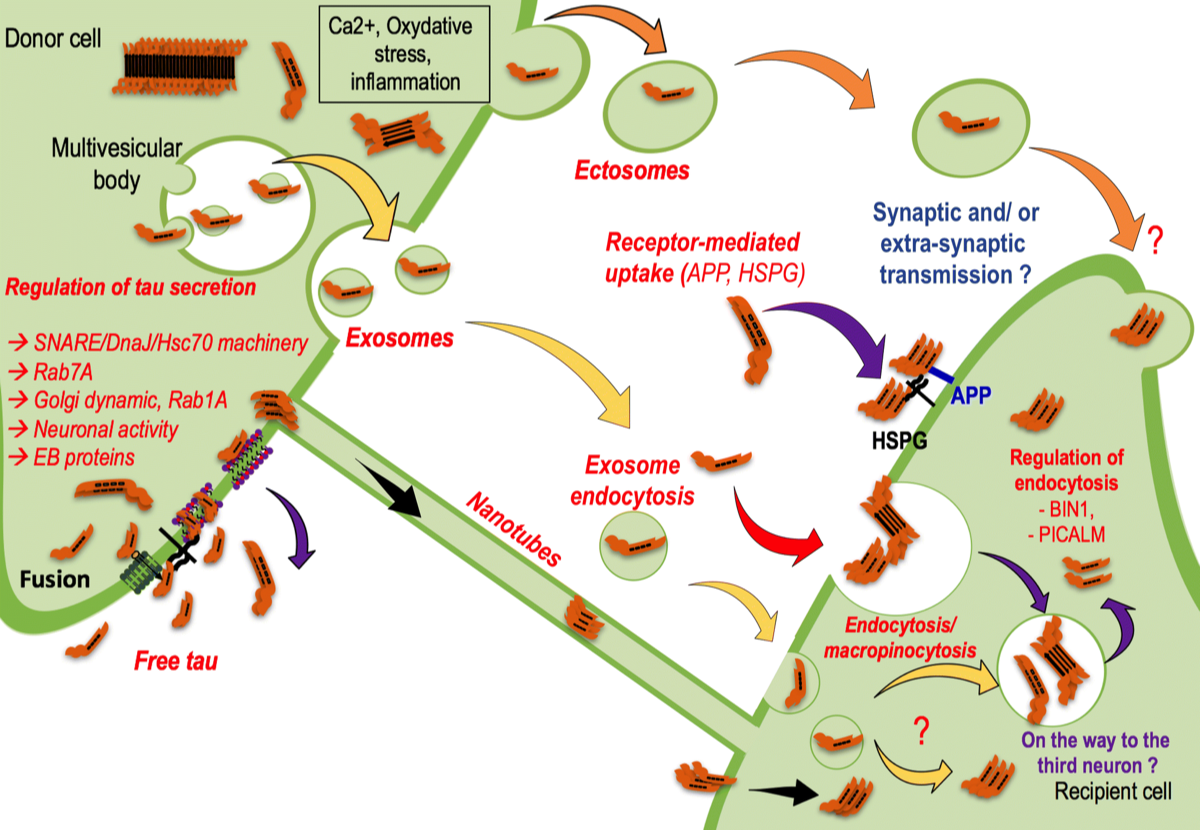
(Colin M. et al., Acta Neuropath, 2020) {31686182}
ADNI研究では、臨床診断とバイオマーカーによる生体病理学的診断の一致率は約90%前後
ADの病態解明に伴い、NIA-AAの研究フレームワークでは、ATNシステムと呼ばれるバイオマーカーに基づく新しい診断法が提唱されている{29653606}。髄液のアミロイドβ1-42の低下、あるいはアミロイドPETによる大脳皮質へのリガンド結合はアミロイド斑のバイオマーカー(A)となる。髄液のリン酸化タウの上昇、タウPETでリガンド結合は繊維性タウのバイオマーカー(T)となる。髄液の総タウの上昇、FDG PETの代謝低下、MRIでの脳萎縮は、神経変性または神経細胞傷害のバイオマーカー(N)である。このシステムによると、認知症の有無に関わらす細胞外のアミロイド斑(A)と細胞内の神経原線維変化(T)がADの病理組織学的特徴となる。
興味深いことに、ADNI研究で臨床診断されたADの90%程度が、髄液バイオマーカーを用いて評価されたin vivo pathologyの所見と一致した{31914216}{DOI: 10.4172/2161-0460.1000021}{19539742}。この一致率は他のコホートの研究でも同レベルである{22170879}。Consortium to Establish a Registry for Alzheimer's Disease (CERAD)研究の報告では、臨床診断されたAD患者の87%が、剖検で神経病理学的に確認されると報告されている{7898697}。
現時点ではADの病態をターゲットにした治療法は開発されていないが、今後はこれらのバイオマーカーの結果に応じて治療法が選択されることになると思われる。最近報告された221AD301 ENGAGE試験では、主要評価項目であるClinical Dementia Rating(CDR)のSum of Boxesについて、アデュカヌマブの最高用量を投与された被験者の認知機能の低下が有意に抑制された可能性が報告された。対象となったのは、アミロイドPET陽性の軽度のADまたはMCI患者で、MMSEスコアは24~30の範囲。本研究では、凝集したアミロイドβのクリアランスがAD病態による認知機能低下を抑制することが示唆されたこと、そして何よりも、MCI、さらには早期のADにおいてもdisease modifying therapy (DMT)が有効であることが注目される。興味深いことにアミロイドとタウのPETによる前向き研究では、臨床的に認知機能が正常な高齢者の脳において、先行するアミロイドβの上昇が後続するタウの変化と関連することが示されたていることから、さらに早い段階で治療を開始した方が効果的であると思われる。しかしながらアミロイドβ沈着のある人が必ずしもADに進行するわけではないことにも注意が必要で、どのタイミングで治療を開始するのが効率的であるかを考慮する必要がある。例えば、NIA-AAの基準によると、アミロイド病理をもつ人の進行リスクはわずか22%であった。したがって、すべてのアミロイドβ陽性MCI患者がアミロイドβ修飾療法の決定的な候補となるわけではない。先行研究では、アミロイドβ陽性MCI患者の転換率は40%~71%と報告されている。これらの研究間のばらつきは、追跡期間よりもむしろ被験者のアミロイドβおよびタウ病理の進行速度に依存しているとも考えられる。
専門医のための認知症テキスト
認知症関連ページ