6. 前頭側頭型認知症(FTD)、前頭側頭葉変性症(FTLD)
アルツハイマー病が知覚系大脳皮質の病変を主体とするのに対し、運動系大脳皮質病変を主体とする非アルツハイマー型の変性型認知症疾患を包括的に捉えた疾患群。原因としてタウの変性、TDP-43やFUSが関係しているものが知られている。TDP-43とFUSが関係しているものは、運動障害(筋萎縮性側索硬化症)を伴うもの(FTLD-MND)と伴わないもの(FTLD-nonMND)に分けられる。タウの変性が原因となるものは、3Rタウの変性が主体であるPick病、4Rタウの変性が主体である疾患群(皮質基底核変性症、進行性核上性麻痺、嗜銀顆粒性認知症)に分類される。
前頭側頭型認知症の発症には、遺伝的背景が強い(40~50%)ためか、多くは65歳以下とアルツハイマー病よりも若く発症し、65歳以下の認知症ではアルツハイマー病に次いで多い。記憶の障害は目立たたず、異常な言動や行動を主訴に家族が相談に来る場合が多い。前頭側頭型認知症の原因がタウかTDP-43かを病理以外の検査で判別するのは難しいため、臨床上は症状(障害部位)によって行動障害型認知症(bv-FTD)と原発性進行性失語症(PPA)に分類することが多い。PPAはさらに意味性認知症(SD)、進行性非流暢性失語症(PNFA)に分類され、これらをまとめて前頭側頭葉変性症(FTLD)と呼ぶ。
FTLDは関連する蛋白によって、FTLD-tau、FTLD-TDP、FTLD-FUSに分類される。FTLDを原因ではなく障害部位(症状)で分類すると、前頭側頭型認知症(frontotemporal dementia: FTD)、意味性認知症(semantic dementia:SD)、進行性非流暢性失語症(progressive non-fluent aphasia:PNFA)に分類される。一方、FTDは行動障害型(behavioral variant:bvFTD)、非流暢/失文法型PPA(non-fluent variant:nfv PPA)、意味型 PPA(semantic variant:svPPA)に分類される。 紛らわしいのは「FTD」という記載がbvFTDを意味しているのか、PPAも含めているのかで、FTLDの中にbvFTD、PNFA、SDがあると考えておいたほうがすっきりする。なお、bvFTDは症状からさらに脱抑制型、無欲型、常同型に分類される。

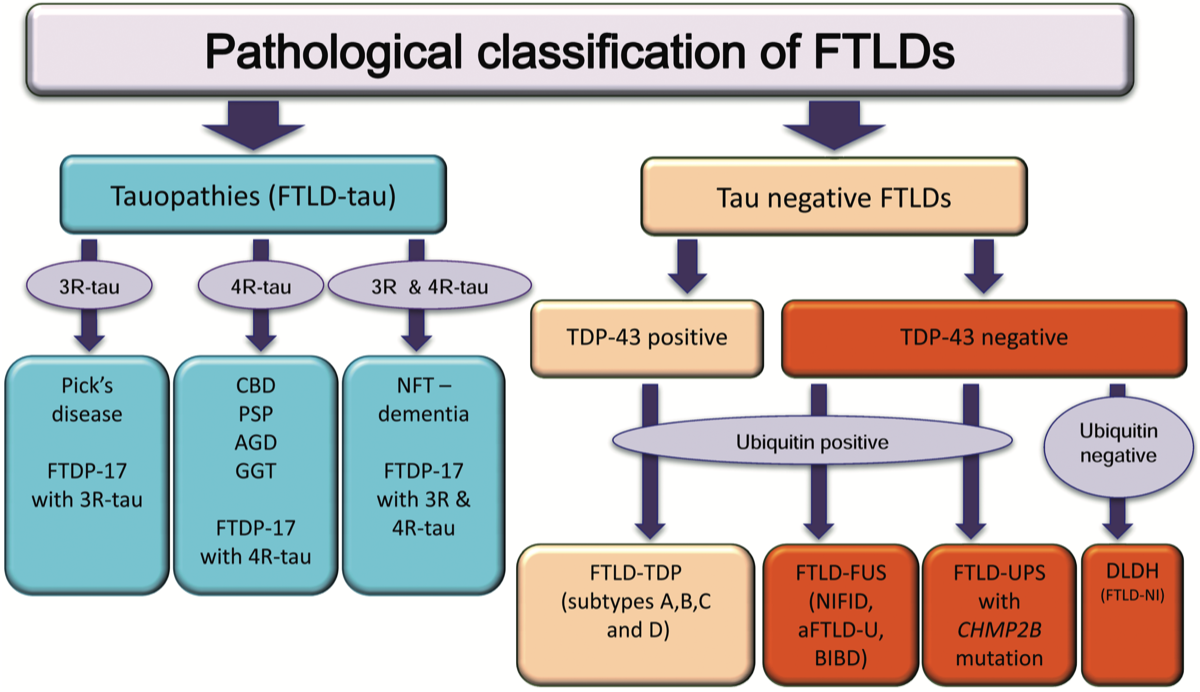
aFTLD-U, atypical frontotemporal lobar degeneration with ubiquitin immunoreactive neuronal inclusions; AGD, argyrophilic grain disease; BIBD, basophilic inclusion body disease; CBD, corticobasal degeneration; DLDH, dementia lacking distinctive histology; FTLD, frontotemporal lobar degeneration; FTDP-17, frontotemporal dementia and parkinsonism linked to chromosome 17; GGT, globular glial tauopathy, NFT-dementia, neurofibrillary tangle dementia; NIFID, neuronal intermediate filament inclusion disease. (Lashley T. et al., Neuropathol Appl Neurobiol, 2015) {26041104}
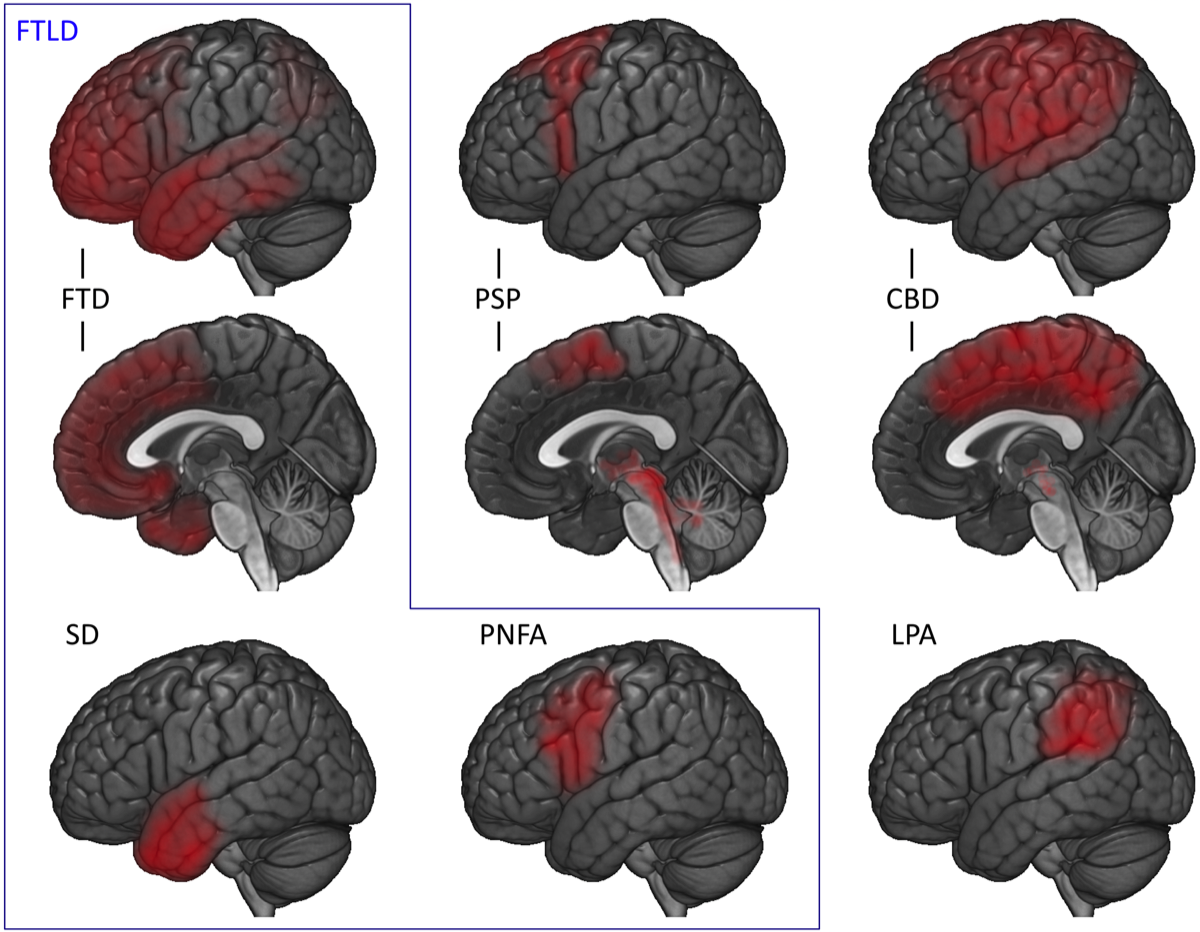
MRIや脳血流SPECTで前頭葉や側頭葉を中心とした萎縮や脳血流低下が認められ、心理検査でADよりも前頭葉の機能低下が目立つ。典型的には図のような領域の血流低下と脳萎縮が認められる。
障害部位(症状)による分類を図に示した。この図でロゴペニック型 PPA(logopenic variant PPA:lvLPA)は、優位半球の頭頂葉の障害で、ADの病態が原因であることが多いと考えられる。なお、FTLD-tauには3R tauopathyであるPick病が含まれる。
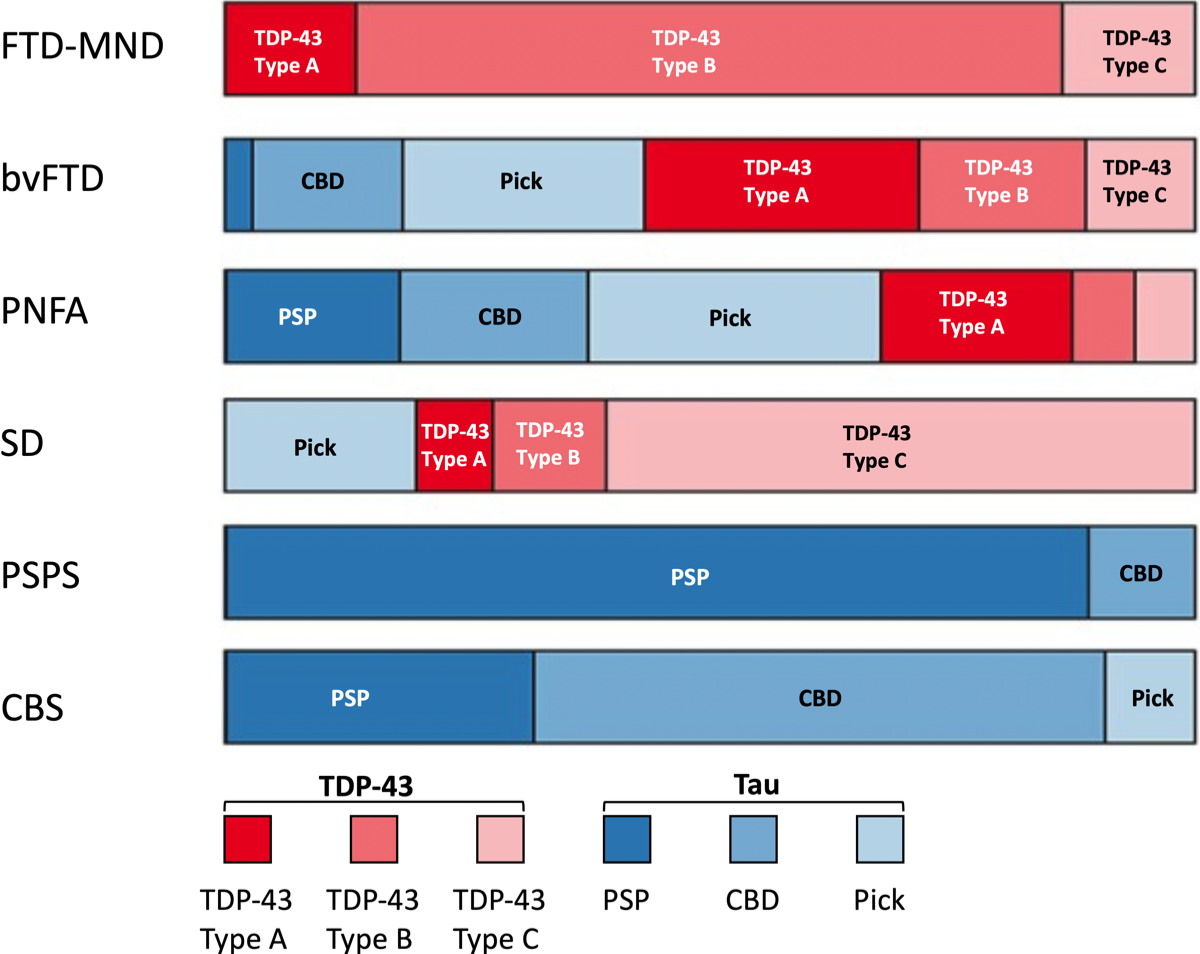
SDとFTD-MNDはほとんどがFTLD-TDPであるのに対し、PSPSPSとCBSはほとんどがタウ病理を特徴としている。(D’Alton S. et al., Front Aging Neurosci, 2014) {25191265}
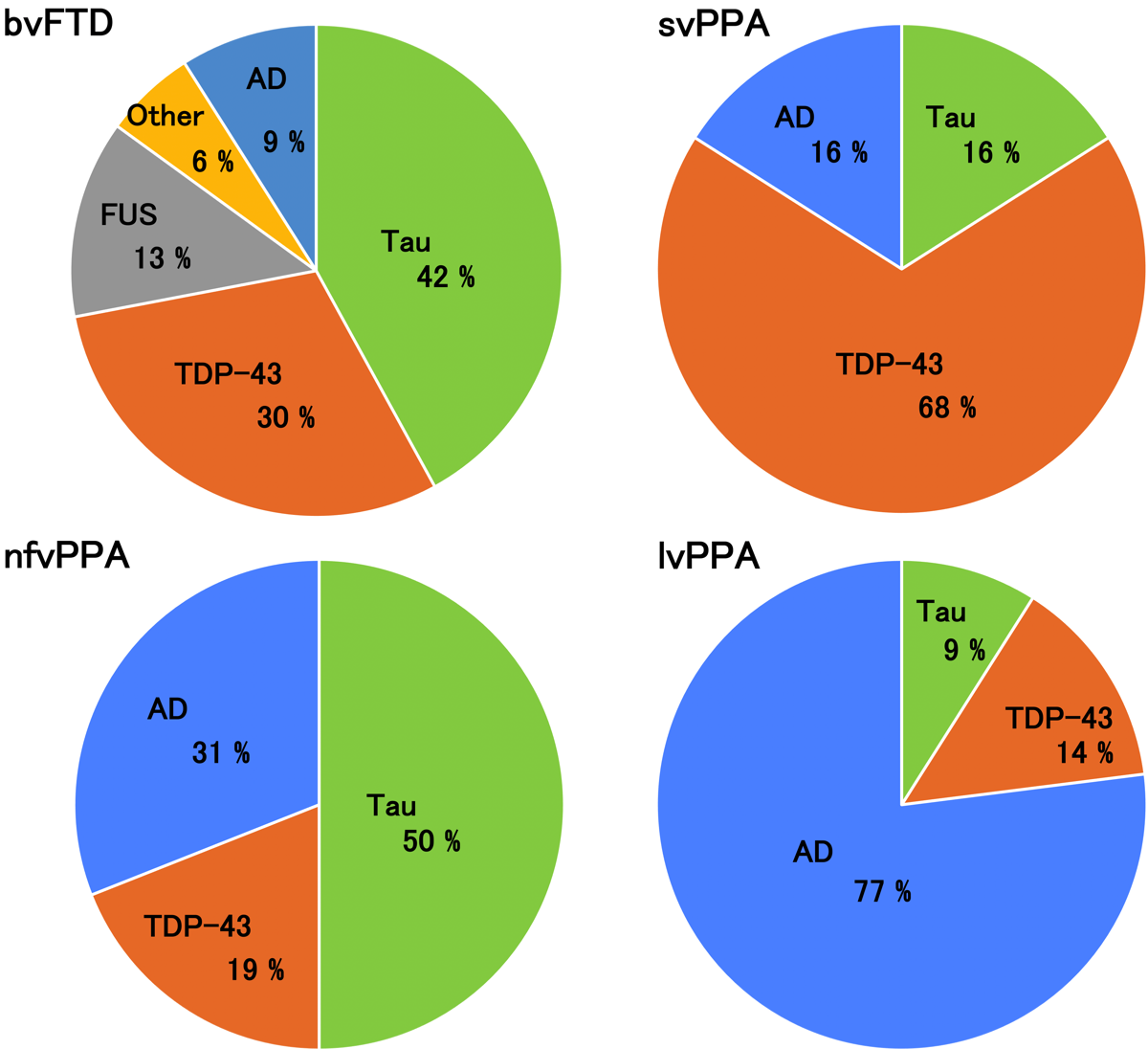
FTLDの原因別頻度を示す。bvFTDの原因は、タウとTDP-43が多く、ついでFUS、ADとなる。PPAの中でsvPPAはTDP-43、nfvPPAはタウ、lv-PPAはADが多い。どのサブタイプも男性に多く、発症年齢は50代から60代でbvFTDの発症年齢はこれらの中でも若い傾向が認めらた。どのタイプも重複した症状を呈するが、bvFTDは常同行動、svPPAは語想起障害や語義失語と表層失読、nfv-PPAは発語失行、logopenic PPA(lvPPA)では語想起障害が特徴的とされている。
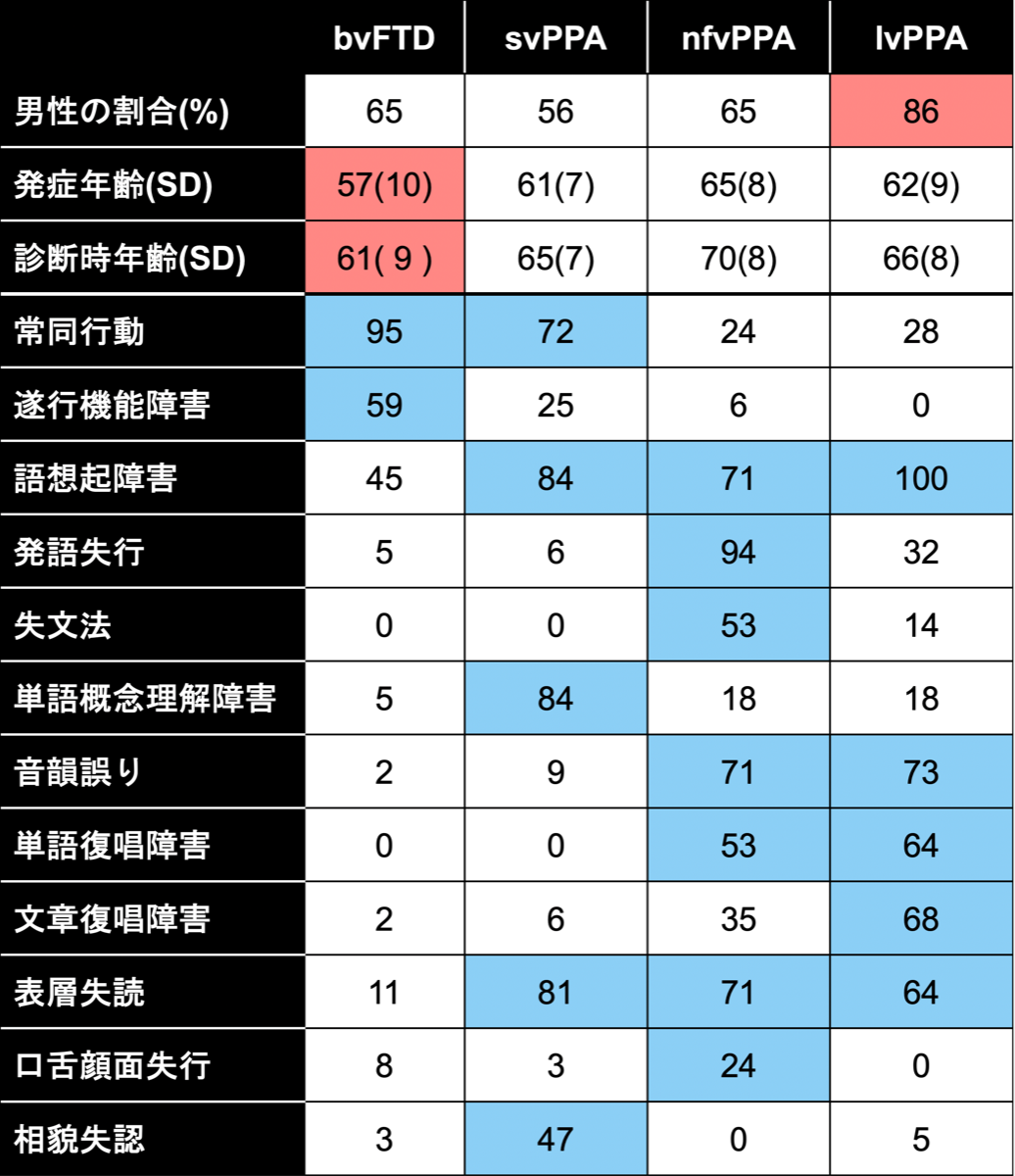
(Chare L., et al., JNNP, 2014) {24421286}
次のような症状が特徴。
自発性の低下:前頭葉内側障害の症状と考えられ、じっとしているかと思うと常同的な行動を示すことがあり、抑うつ感や悲哀、不安感は目立他ない。寝てばかりいるので声をかけても、「何か食べる?」「知らん」、「散歩する?」「知らん」と何も考えずに即答する傾向がある。
常同行動:同じ言葉を意味もなく繰り返す、同じところを同じ時間に周遊する、など日常の行動のパターンにバリエーションがなくなってくる。ささいな事にこだわる傾向あり。前頭葉底面の障害と考えられる。
遂行機能障害:例えば、料理のレシピ考えて、買い物をして、作る順番やタイミングを考えて調理するといった複合的な行動が完遂できない。
問題行動:周りのことを気にせず自分の思うがままに行動する。周囲の人の気持ちは一切意に介さず、共感することもない。注意されると激しく怒り、ルールは無視して、万引きなどの反社会的な行動をしても反省するそぶりは示さない。
鑑別すべき疾患に、その他の変性型認知症、血管性認知症、遅発性統合失調症、うつ病、発達障害がある。うつ病では悲哀感、不安、罪責感があるが、bvFTDでは感覚鈍麻、アパシーが主体となる。初老期の女性に多く認められる遅発緊張病は、抑欝、不安・焦燥、幻覚・妄想、緊張病症候群(情緒不安定、拒絶、昏迷、自律神経症状)が主な症状。遅発性統合失調症(妄想性障害)は頑固な妄想を主体とした症状で、bvFTDのように人格の崩壊はなく、妄想以外に異常は目立たない。幻聴を訴えることが多い。初老期以降に顕在化してくる発達障害は、加齢とともに脱抑制が目立つようになる。この場合の発達障害とは、知的レベルというよりも自閉症スペクトラム障害(ASD; autism spectrum disorder)やアスペルガー症候群といったパーソナリティーにかかわる問題であり、高学歴の症例であってもbvFTDと鑑別を要する。万引きを繰り返す、ギャンブルにはまる、ストーカー行為を繰り返す、対人とのトラブルを頻回におこすなどの行動異常があり、bvFTDとの鑑別に苦慮することがある。若い頃から交友関係が少ない、怒りっぽい、衝動的な傾向がある、MRIやSPECTで前頭葉に器質的な異常が見られないことなどが参考になる。

Ⅰ. 神経変性疾患
行動/認知機能障害が緩徐進行性である
Ⅱ. Possible bvFTD(A~Fの3項目以上)
A. 発症3年以内に認められる脱抑制行動
1.社会的に不適切な行動
2.礼儀やマナ-の欠如
B. 発症3年以内に認められるアパシ-または無気力
1.アパシ-(無関心)
2.無気力
C. 発症3年以内に認められる共感(sympathy)や感情移入(empathy)の欠如
1.他人が求めることや感情を意に介さない
2.社会的関心、相互関係、人間的な温かさの欠如
D. 発症3年以内に認められる保続的、常同的、強迫的/儀式的行動
1.単純動作の反復(タッピング、スクラッチング、つまむ、唇ならし、など)
2.複雑な動作の強迫的、儀式的行動(カウントやクリ-ニングの儀式、同じものを集める、必要もないのに同じ場所に行く、など)
3.常同言語(コミュニケ-ションとは関係なく、同じ単語やフレ-ズ、物語を繰り返す)
E. 口唇傾向、食習慣の変化
1.食事嗜好の変化
2.過食や飲酒、喫煙の増加
3.何でも口に入れる傾向(口唇的探索、異食)
F. 神経心理学的検査において、記憶や視空間認知機能は比較的保持されているにもかかわらず、遂行機能障害が認められる。
1.遂行機能障害
2.経験記憶障害は比較的保たれる
3.視覚空間機能は比較的保たれる
Ⅲ. Probable bvFTD
A. Possible bvFTD の条件を満たす
B. 明らかな認知機能低下(介護者またはCDRやFunctional Activities Questionnaire scoreの悪化)
C. 画像検査でbvFTDに矛盾しない結果
1.前頭葉や側頭葉前部の萎縮(CT、MRI)
2.前頭葉や側頭葉前部の血流低下や代謝低下(PET、SPECT)
Ⅳ. 病理所見を伴う Definite bvFTD
A. Probable bvFTD または Possible bvFTD の条件を満たす
B. 病理でFTLD 所見(生検または剖検)
C. 判明しているFTLD の遺伝子変異
Ⅴ. 除外項目
以下のAとBは陰性、Cはpossible bvFTDでは陽性でもかまわないが、probable bvFTDでは陰性。
A. その他の非変性神経疾患や内科的疾患の可能性(せん妄、虚血疾患、外傷、感染、甲状腺機能低下症、栄養失調など)
B. 異常行動が精神疾患の可能性
C. バイオマ-カ-検査が、アルツハイマ-病やその他の神経変性疾患を示唆
性格変化と社会的行動の障害(disorderedsocialconduct)が、発症から疾 患の経過を通して優位な特徴である。知覚、空間的能力、行為、記憶といった道具的認知機能は正常か、比較的良好に保たれる。
Ⅰ. 主要診断特徴(すべて必要)
A. 潜行性の発症と緩徐な進行
B. 社会的対人行動(interpersonalconduct)の早期からの障害
C. 早期からの自己行動の統制(regulationofpersonalconduct)障害
D. 早期からの情意鈍麻(emotionalblunting) E.早期からの病識の欠如
Ⅱ. 支持的診断特徴
A. 行動異常
1. 自己の衛生や身なりの障害
2. 精神の硬直化と柔軟性のなさ
3. 易転導性(distractibility)と維持困難(impersistence)
4. 口唇傾向と食餌嗜好の変化
5. 保続的行動と常同行動
6. 使用行動
B. 音声と言語
1.音声出力の変化
a. Aspontaneity and economy
(自分から会話を始めない、短いフレーズに限られる)
b. Press of speech (途切れ途切れに話し、会話を独り占めする)
2. 言語のステレオタイプ化
3. 反響言語 (echolalia)
4. 保続
5. 寡黙(mutism)
Ⅲ. FTLDに共通する支持的診断特徴
A. 65歳以前の発症.親兄弟に同症の家族歴
B. 球麻痺,筋力低下と萎縮,筋線維束攣縮.保続的行動と常同行動
Ⅰ. 神経変性疾患
行動/認知機能障害が緩徐進行性である
Ⅱ. Possible bvFTD(A~Fの3項目以上)
A. 発症3年以内に認められる脱抑制行動
1.社会的に不適切な行動
2.礼儀やマナ-の欠如
B. 発症3年以内に認められるアパシ-または無気力
1.アパシ-(無関心)
2.無気力
C. 発症3年以内に認められる共感(sympathy)や感情移入(empathy)の欠如
1.他人が求めることや感情を意に介さない
2.社会的関心、相互関係、人間的な温かさの欠如
D. 発症3年以内に認められる保続的、常同的、強迫的/儀式的行動
1.単純動作の反復(タッピング、スクラッチング、つまむ、唇ならし、など)
2.複雑な動作の強迫的、儀式的行動(カウントやクリ-ニングの儀式、同じものを集める、必要もないのに同じ場所に行く、など)
3.常同言語(コミュニケ-ションとは関係なく、同じ単語やフレ-ズ、物語を繰り返す)
E. 口唇傾向、食習慣の変化
1.食事嗜好の変化
2.過食や飲酒、喫煙の増加
3.何でも口に入れる傾向(口唇的探索、異食)
F. 神経心理学的検査において、記憶や視空間認知機能は比較的保持されているにもかかわらず、遂行機能障害が認められる。
1.遂行機能障害
2.経験記憶障害は比較的保たれる
3.視覚空間機能は比較的保たれる
Ⅲ. Probable bvFTD
A. Possible bvFTD の条件を満たす
B. 明らかな認知機能低下(介護者またはCDRやFunctional Activities Questionnaire scoreの悪化)
C. 画像検査でbvFTDに矛盾しない結果
1.前頭葉や側頭葉前部の萎縮(CT、MRI)
2.前頭葉や側頭葉前部の血流低下や代謝低下(PET、SPECT)
Ⅳ. 病理所見を伴う Definite bvFTD
A. Probable bvFTD または Possible bvFTD の条件を満たす
B. 病理でFTLD 所見(生検または剖検)
C. 判明しているFTLD の遺伝子変異
Ⅴ. 除外項目
以下のAとBは陰性、Cはpossible bvFTDでは陽性でもかまわないが、probable bvFTDでは陰性。
A. その他の非変性神経疾患や内科的疾患の可能性(せん妄、虚血疾患、外傷、感染、甲状腺機能低下症、栄養失調など)
B. 異常行動が精神疾患の可能性
C. バイオマ-カ-検査が、アルツハイマ-病やその他の神経変性疾患を示唆
性格変化と社会的行動の障害(disorderedsocialconduct)が、発症から疾 患の経過を通して優位な特徴である。知覚、空間的能力、行為、記憶といった道具的認知機能は正常か、比較的良好に保たれる。
Ⅰ. 主要診断特徴(すべて必要)
A. 潜行性の発症と緩徐な進行
B. 社会的対人行動(interpersonal conduct)の早期からの障害
C. 早期からの自己行動の統制(regulation of personalconduct)障害
D. 早期からの情意鈍麻(emotional blunting)
E. 早期からの病識の欠如
Ⅱ. 支持的診断特徴
A. 行動異常
1. 自己の衛生や身なりの障害
2. 精神の硬直化と柔軟性のなさ
3. 易転導性(distractibility)と維持困難(impersistence)
4. 口唇傾向と食餌嗜好の変化
5. 保続的行動と常同行動
6. 使用行動
B. 音声と言語
1.音声出力の変化
a. Aspontaneity and economy
(自分から会話を始めない、短いフレーズに限られる)
b. Press of speech (途切れ途切れに話し、会話を独り占めする)
2. 言語のステレオタイプ化
3. 反響言語 (echolalia)
4. 保続
5. 寡黙(mutism)
Ⅲ. FTLDに共通する支持的診断特徴
A. 65歳以前の発症.親兄弟に同症の家族歴
B. 球麻痺,筋力低下と萎縮,筋線維束攣縮.保続的行動と常同行動
意味記憶(semantic memory)は体験記憶と異なり、自分の経験した事とは直接関わりはないが概念的知識として記憶していることをさす。見たもの(特定の景色や相貌)や聞いたもの(特定の言葉)を解釈して概念化する作業には前頭葉が使われるが、側頭葉の前部は前頭葉によるこうした概念を言語(画像)と関連づけをしている。このため側頭葉の前部が障害されると、物事の法則や概念などの記憶(知識)が障害さる。
語の意味(概念)が障害されている状態は語義失語と呼ばれ、理解を伴わない書き取りや復唱は可能で、語性錯語はあるが流暢に話すことは可能。語義失語は左(優位半球)の側頭葉先端部の障害で起きやすく、右の場合は視覚的情報の意味や概念の障害が主体となる。
具体的には以下のような症状として認められる。
物品呼称の障害(語想起障害)
単語理解の障害
物事の言語的な解釈の障害(語義失語)
対象物に対する知識の障害
有名人や友人、たまにしか会わない親戚の顔が認識できない
金閣寺やスカイツリーを見ても何であるか認知できない
表層性失読・失書 (例外的な規則の単語が読めない、書けない)
意味性認知症の診断基準(難病情報センタ-)
Ⅰ. 必須項目:次の2つの中核症状の両者を満たし、それらにより日常生活が阻害されているa)
A.物品呼称の障害
B.単語理解の障害
Ⅱ. 以下の 4 つのうち少なくとも3つを認める
1.対象物に対する知識の障害b)(特に、低頻度/低親密性のもので顕著)
2.表層性失読・失書c)
3.復唱は保たれ、流暢性の発語を呈する
4.発話(文法や自発語)は保たれる
Ⅲ. 高齢発症も存在するが、70 歳以上では稀
Ⅳ. 画像検査:前方優位の側頭葉にMRI/CTでの萎縮がみられる
Ⅴ. 除外診断:以下の疾患を鑑別できる
1)アルツハイマ-病
2)レビ-小体型認知症
3)血管性認知症
4)進行性核上性麻痺
5)大脳皮質基底核変性症
6)うつ病などの精神疾患
臨床診断: I ~ Vのすべてを満たすもの
注1)高齢の発症は少なく、発症年齢65歳以下を対象とする
注2)画像読影レポ-トまたはそれと同内容の文書の写し(判読医の氏名の記載されたもの)を添付すること。なお、画像検査所見および除外診断については、別表を参考に鑑別を行う。
注3)特徴的な言語の障害に対して、本人や介護者はしばしば“物忘れ”として訴えることに留意する
注4)bvFTDと同様の行動障害がしばしばみられることに留意する
a)例: これらの障害に一貫性がみられる。つまり、異なる検査場面や日常生活でも同じ物品、単語に障害を示す。
b)例: 富士山や金閣寺の写真を見せても、山や寺ということは理解できても特定の山や寺と認識できない。
信号機を提示しても「信号機」と呼称ができず、「見たことない」、「青い電気がついとるな」などと答えたりする。
有名人や友人、たまにしか会わない親戚の顔が認識できない。それらを見ても、「何も思い出せない」、「知らない」と言ったりする。
c)例: 団子→“だんし”、三日月→“さんかづき”
語義失語の症状
1. 語義の障害
言葉の意味がわからない: 動物の名前を挙げてください。「動物って何?」
2. 物品呼称・同定の障害(語想起障害、喚語困難、健忘失語)
物の名前が出てこない: 鉛筆を見て、「紙に書く物で、何と言ったっけ?」。
語頭音(手がかり)の効果なし: ヒントで“エンピ….”と言うと、「エンピ と言うんですか?」
3. ことわざの補完障害
「サルも木から….」「犬も歩けば….」「弘法も….」
4. 表層性失読・失書
不規則な読みが必要な単語が読めない、書けない: 三味線、海老、爪楊枝、 団子、三日月….
5. 復唱は可能、経験記憶は保たれる。
PNFAは非流暢/失文法型PPA (non fluent/agrammatic variant PPA)とも呼ばれる。構音にかかわる一次運動野からその高次の発語にかかわる前頭前野(運動性言語中枢、Broca's area)の障害(従って優位半球側)によって出現する。Broca失語あるいは超皮質性運動失語としての症状、口部顔面失行(前弁蓋部症候群)として嚥下障害を伴うものなどがある。
自発語の減少(言葉が出にくいと訴えることが多い) 、流暢に話すことができず努力性 、外国語-片言しゃべり、失構音(発語失行) などの症状がみられる。発語失行の場合、下位脳神経麻痺による構音障害と異なり構音の障害のパターンが不規則に出現する。具体的には、音の連結不良(言葉の出だしが困難)、不規則な構音の歪み、プロソディ障害(抑揚や速度、リズムの乱れ)などとしてみられる。
nfvPPAの診断基準 (Gorno-Tempini) 21325651
Ⅰ. 次の中核症状のうち少なくとも1つが存在
A.発話時の失文法
B.一貫しない音の誤りと歪みを伴う努力性のたどたどしい発語(発語失行)
Ⅱ. 次の3つの特徴のうち少なくとも2つを満たす
A.構文が複雑だと理解できない
B.単語レベルでの理解は保たれている
C.対象物の知識は保たれている
優位半球頭頂葉の障害によるためFTLDの範疇には入らないが、原発性進行性失語症(PPA)に分類される。基礎疾患はADのことが多いが、タウの病変やTDPの病変でも起こる。"logopenic"とは語="logo"と少ない="penic"の意味で、自発語および呼称における単語想起障害があり、超皮質性失語症と異なり復唱障害も認められる。
lvPPAの診断基準 (Gorno-Tempini) 21325651
Ⅰ. 次の次の中核症状のうち少なくとも1つが存在
A.自発話および呼称における単語想起障害
B.フレ-ズや文章の復唱障害
Ⅱ. 次の3つの特徴のうち少なくとも3つを満たす
A.自発話および呼称における発話の音韻の誤り
B.単語理解と対象物の知識は保たれている
C.運動性失語はない
D.明らかな失文法はない
Transactive response (TAR) DNA-binding protein of 43 kDa (TDP-43)は核内に多く存在し、後述のFUSと同様にRNA-binding protein (RBPs) の一種である。核内でpre-mRNA、microRNAs、long noncoding RNAsのsplicingや生成制御、細胞質内でmRNAの安定化や輸送、翻訳に関係している。正常な神経細胞では核内に多く存在するが、病気になると細胞質に多く出現するようになり、やがて凝集して封入体が形成される。FTLDの50%以上、sporadic ALSの95%にTDP-43の封入体が認められる。またADの50%以上にもTDP-43の病変が認められ、加齢に伴う海馬硬化でも出現する。後述のLimbic-predominant age-related TDP-43 encephalopathy (LATE)は、超高齢者でADに似た脳萎縮を呈してくる。
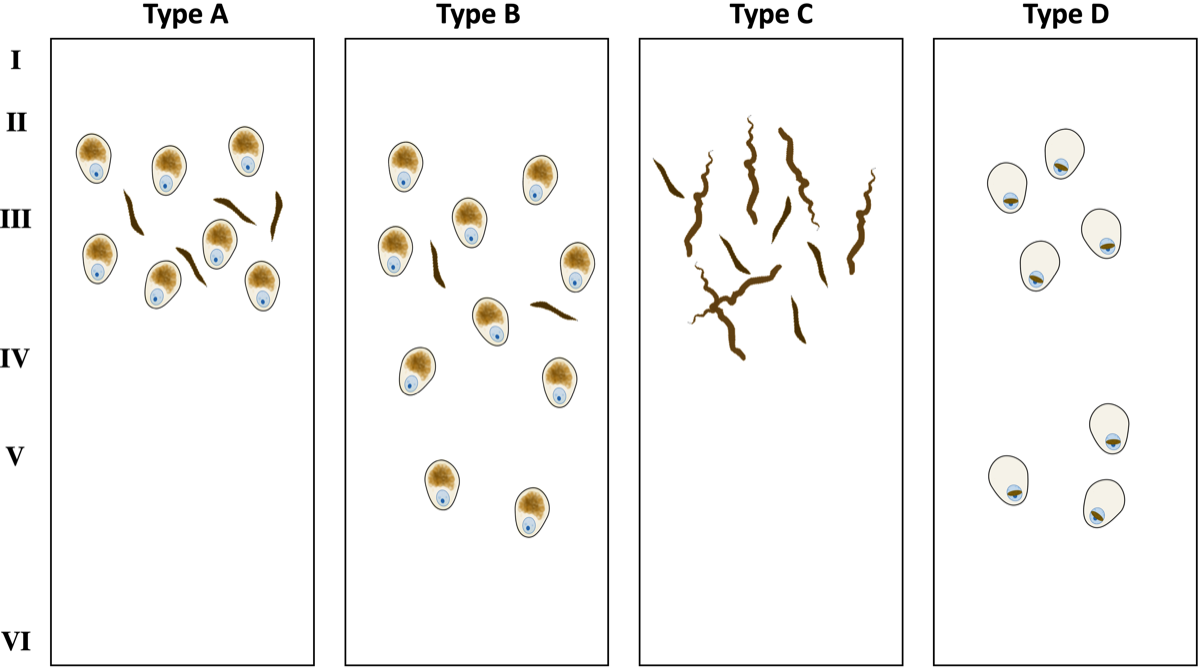
病理学的にTDP-43疾患は4タイプに分類される。TDP-43が陽性となるものに、neuronal cytoplasmic inclusions (NCIs)、dystrophic neurites (DNs)があり、NCIsとshort DNsが大脳皮質表層(layers II/III)にあるものをtype A、NCIsが全層に認められるものをtype B、long DNsが表層に認められるものを type C、核内にレンズ型の封入体(neuronal intranuclear inclusions, NII)を認めるものを type Dに分類する。Type Aは左右非対称で萎縮が強く、Type Bは左右対照的な萎縮で側頭葉の萎縮は軽度なことが多い。
Transactive response (TAR) DNA-binding protein of 43 kDa (TDP-43)は核内に多く存在し、後述のFUSと同様にRNA-binding protein (RBPs) の一種である。核内でpre-mRNA、microRNAs、long noncoding RNAsのsplicingや生成制御、細胞質内でmRNAの安定化や輸送、翻訳に関係している。正常な神経細胞では核内に多く存在するが、病気になると細胞質に多く出現するようになり、やがて凝集して封入体が形成される。FTLDの50%以上、sporadic ALSの95%にTDP-43の封入体が認められる。またADの50%以上にもTDP-43の病変が認められ、加齢に伴う海馬硬化でも出現する。
病理学的にTDP-43が関係するFTLDは4タイプに分類される。病理標本でTDP-43が陽性となるものに、neuronal cytoplasmic inclusions (NCIs)、dystrophic neurites (DNs)があり、NCIsとshort DNsが大脳皮質表層(layers II/III)にあるものをtype A、NCIsが全層に認められるものをtype B、long DNsが表層に認められるものを type C、核内にレンズ型の封入体(neuronal intranuclear inclusions, NII)を認めるものを type Dに分類する。Type Aは左右非対称で萎縮が強く、Type Bは左右対照的な萎縮で側頭葉の萎縮は軽度なことが多い。
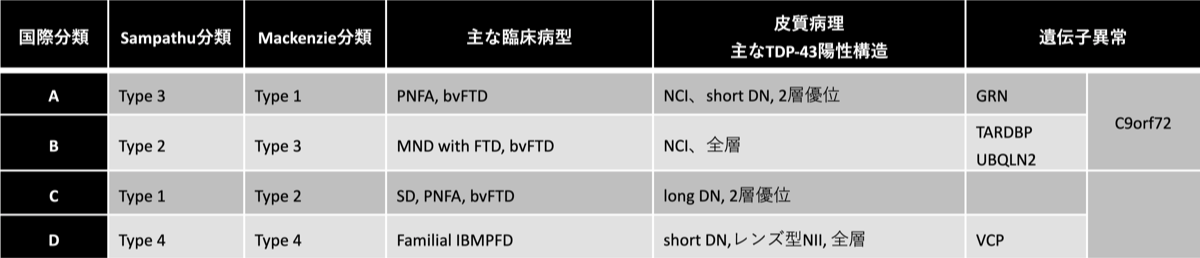
bvFTD: behavioral variant frontotemporal dementia、MND: motor neuron disease(運動ニュ-ロン疾患)、PNFA: progressive non-fluent aphasia(進行性非流暢性失語)、SD: semantic dementia(意味性認知症)、IBMPFD: inclusion body myopathy associated with Paget’s disease of bone and/or frontotemporal dementia(骨ペ-ジェット病と前頭側頭型認知症をともなう封入体筋炎)
C9orf72: chromosome 9 open reading frame 72、TARDBP: Transactive DNA-binding protein、PGRN: progranulin、VCP; valosin-containing protein、UBQLN2: ubiquilin2
NCI: neuronal cytoplasmic inclusion(神経細胞質内封入体)、DN: dystrophic neurites(変性神経突起)
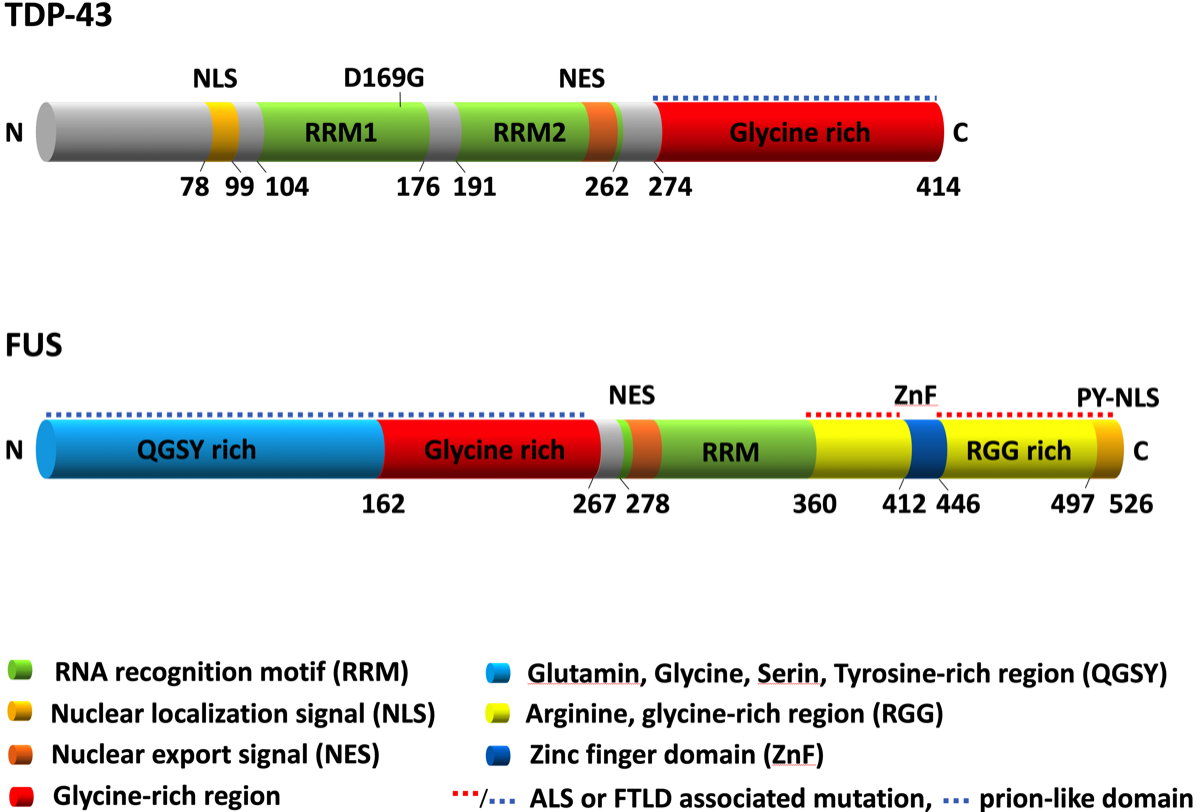
TDP-43の構造と機能
TDP-43はmRNAの管理にかかわる不均一核内リボ核酸蛋白の1種であり、対象は全体の約30%のmRNAであり、細胞の蛋白合成の制御やストレス顆粒の形成などに関わっている。質量が43kDaのRNA/DNA結合蛋白でTARDBP遺伝子によって支配されている。HIV-1 virusのRNAが逆転写され宿主のDNAに取り込まれたプロウイルスの状態で、DNAのTAR領域に結合する蛋白としてTDP-43が発見された。TAR(trans-activation response element)は、HIV virusのRNAの両端のLTR (long terminal repeat)に存在し、ウイルスの転写(複製)に関与する。現在では、TDP-43はRNAに結合するRRM(RNA recognition motif)を2つもつ、不均一核内リボ核酸蛋白(hnRNP; heterogeneous nuclear ribonucleoproteins)の1種であることがわかっている。
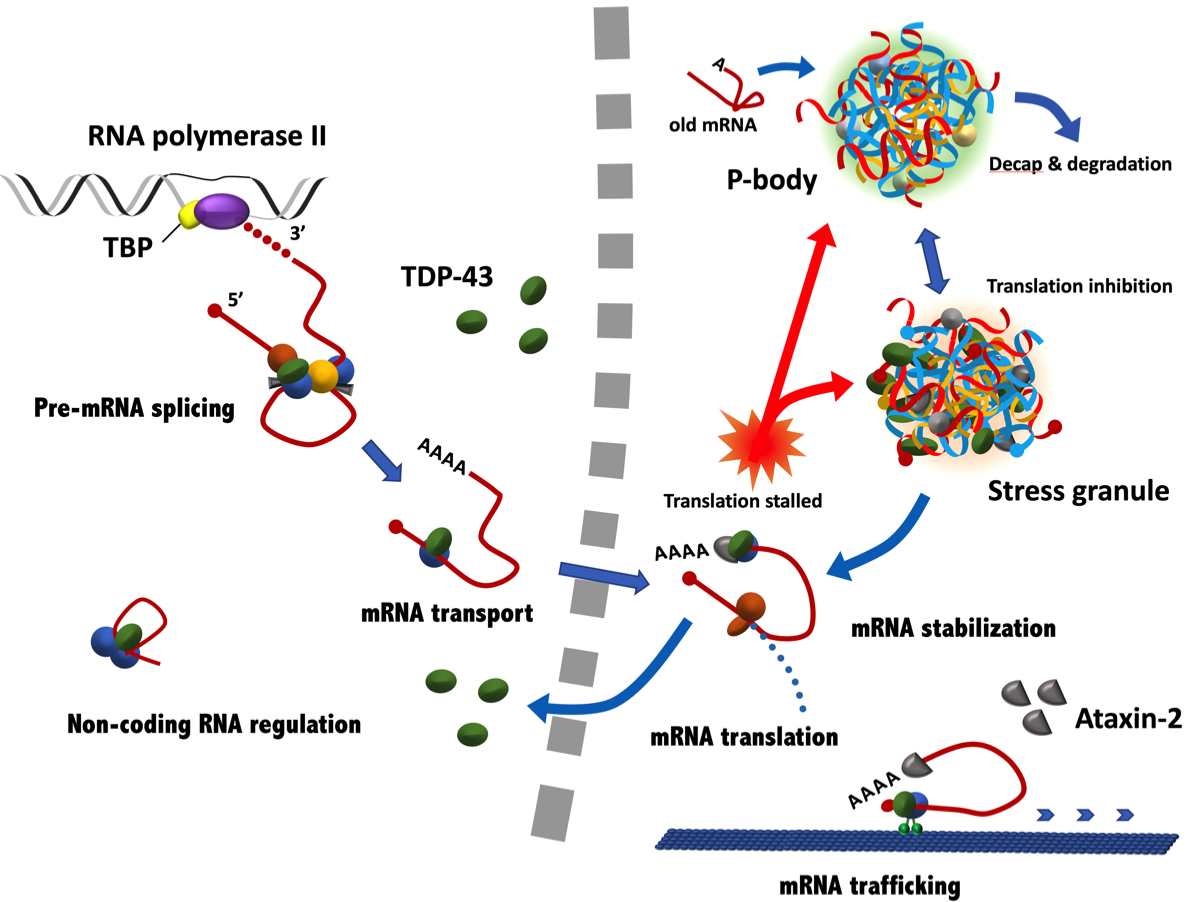
TDP-43をはじめとするhnRNPは、核内で大きなサイズのRNA(heterogeneous nuclear RNA)に結合する蛋白で、DNAの修復やpre-mRNAのスプライシングなどに関与している(下図左)。TDP-43は核外輸送に必要なNES(nuclear export signal)と核内輸送に必要なNLS(nuclear localization sequence)をもち、核と細胞質間をシャトルして遺伝子発現の制御に関わっている(下図右)。興味深いことに、TDP-43は正常ではほとんど核内にとどまっており、ALS/FTDなど病的な状態では細胞質に偏移する。
TDP-43は、核内ではRNA polymeraseで新生されたpre-mRNAに結合して、そのイントロン部分を切除(splicing)して成熟したmRNAの生成に関与する。さらに、mRNAの安定化と核外移送、non-coding RNAs (miRNA、lncRNA、 ncRNA)の安定化にも関与している。TDP-43はUGが豊富な長いイントロンをもつRNA sequenceを好み、対象となるmRNAは少なくとも6000種類、全体の30%にも及ぶ。細胞質内におけるTDP-43の機能は、mRNAからの蛋白合成(translation)、mRNAの安定化と軸索や樹状突起への輸送に関与している(下図左)。TDP-43の生成は、TARDBP遺伝子からつくられた自身のmRNAの3’側のUTR(untranslated region)に結合して厳密に自己制御(autoregulation)されている{21131904}。TDP-43が細胞質で凝集して核に戻る量が少なくなると、負のフィードバックによりTDP-43のm-RNAの産生が亢進する。TDP-43の生産負荷は相対的に不良なTDP-43を増加させ、これが小胞体やリソソームなどに負担をかける可能性がある。
細胞にheat shockなどのストレスが加わると蛋白合成は休止(stalled)するが、TDP-43のようにprion-like domain構造を持つタンパク質は互いに絡み合って、ストレス顆粒(SG; stress granule)を細胞質で形成する。顆粒と言っても膜のような境界はなく、細胞質で液-液相分離(liquid-liquid phase separation)した状態で、mRNAの他にTDP-43を含めたhnRNPs、TIA-1 (T-cell intracytoplasmic antigen; translation repressor)、G3BP1(Ras GAP SH3 domain binding protein 1)、ataxin-2、eIF3、4、5 (eukaryotic initiation factor)、small ribosomal subunitなどが集まっている。ストレスが解消されると分散して蛋白合成が再開されるが、多くはオートファゴゾームで分解さる。なお、異常なmRNAがあると、Dcp、Edc、Patなどの蛋白が集まりP-body(processing body)を形成する{22763747}。この場合、mRNAの5’キャップは取り除かれ(decap)、mRNAは分解される。ストレス顆粒とP-bodyは相互に移行、あるいは両者が合わさったような構造(ドッキング)を取ることが知られている。
詳細は不明であるが、ataxin-2のpolyQ(グルタミン)の繰り返しが27-34程度に延長している場合には、TDP-43によるストレス顆粒の形成が促進されてALSの発症リスクが高くなると推測されている。ちなみにataxin-2のpolyQが34を超えるとSCA2(脊髄小脳変性症2型)を発症することが知られている。アンチセンスオリゴヌクレオチド(ASO)でataxin-2の細胞内生産を抑制することがALSの治療に結びつかないか、研究が行われている。
TDP-43の構造と機能
TDP-43はmRNAの管理にかかわる不均一核内リボ核酸蛋白の1種であり、対象は全体の約30%のmRNAであり、細胞の蛋白合成の制御やストレス顆粒の形成などに関わっている。質量が43kDaのRNA/DNA結合蛋白でTARDBP遺伝子によって支配されている。HIV-1 virusのRNAが逆転写され宿主のDNAに取り込まれたプロウイルスの状態で、DNAのTAR領域に結合する蛋白としてTDP-43が発見された。TAR(trans-activation response element)は、HIV virusのRNAの両端のLTR (long terminal repeat)に存在し、ウイルスの転写(複製)に関与する。現在では、TDP-43はRNAに結合するRRM(RNA recognition motif)を2つもつ、不均一核内リボ核酸蛋白(hnRNP; heterogeneous nuclear ribonucleoproteins)の1種であることがわかっている。
TDP-43をはじめとするhnRNPは、核内で大きなサイズのRNA(heterogeneous nuclear RNA)に結合する蛋白で、DNAの修復やpre-mRNAのスプライシングなどに関与している(下図)。TDP-43は核外輸送に必要なNES(nuclear export signal)と核内輸送に必要なNLS(nuclear localization sequence)をもち、核と細胞質間をシャトルして遺伝子発現の制御に関わっている(下図)。興味深いことに、TDP-43は正常ではほとんど核内にとどまっており、ALS/FTDなど病的な状態では細胞質に偏移する。
TDP-43は、核内ではRNA polymeraseで新生されたpre-mRNAに結合して、そのイントロン部分を切除(splicing)して成熟したmRNAの生成に関与する。さらに、mRNAの安定化と核外移送、non-coding RNAs (miRNA、lncRNA、 ncRNA)の安定化にも関与している。TDP-43はUGが豊富な長いイントロンをもつRNA sequenceを好み、対象となるmRNAは少なくとも6000種類、全体の30%にも及ぶ。細胞質内におけるTDP-43の機能は、mRNAからの蛋白合成(translation)、mRNAの安定化と軸索や樹状突起への輸送に関与している(下図)。TDP-43の生成は、TARDBP遺伝子からつくられた自身のmRNAの3’側のUTR(untranslated region)に結合して厳密に自己制御(autoregulation)されている{21131904}。TDP-43が細胞質で凝集して核に戻る量が少なくなると、負のフィードバックによりTDP-43のm-RNAの産生が亢進する。TDP-43の生産負荷は相対的に不良なTDP-43を増加させ、これが小胞体やリソソームなどに負担をかける可能性がある。
細胞にheat shockなどのストレスが加わると蛋白合成は休止(stalled)するが、TDP-43のようにprion-like domain構造を持つタンパク質は互いに絡み合って、ストレス顆粒(SG; stress granule)を細胞質で形成する。顆粒と言っても膜のような境界はなく、細胞質で液-液相分離(liquid-liquid phase separation)した状態で、mRNAの他にTDP-43を含めたhnRNPs、TIA-1 (T-cell intracytoplasmic antigen; translation repressor)、G3BP1(Ras GAP SH3 domain binding protein 1)、ataxin-2、eIF3、4、5 (eukaryotic initiation factor)、small ribosomal subunitなどが集まっている。ストレスが解消されると分散して蛋白合成が再開されるが、多くはオートファゴゾームで分解さる。なお、異常なmRNAがあると、Dcp、Edc、Patなどの蛋白が集まりP-body(processing body)を形成する{22763747}。この場合、mRNAの5’キャップは取り除かれ(decap)、mRNAは分解される。ストレス顆粒とP-bodyは相互に移行、あるいは両者が合わさったような構造(ドッキング)を取ることが知られている。
詳細は不明であるが、ataxin-2のpolyQ(グルタミン)の繰り返しが27-34程度に延長している場合には、TDP-43によるストレス顆粒の形成が促進されてALSの発症リスクが高くなると推測されている。ちなみにataxin-2のpolyQが34を超えるとSCA2(脊髄小脳変性症2型)を発症することが知られている。アンチセンスオリゴヌクレオチド(ASO)でataxin-2の細胞内生産を抑制することがALSの治療に結びつかないか、研究が行われている。
Stress granule(SG)の形成は、細胞保護的に作用しており、TDP-43はSGの形成を促進する。一方、FUSはDNAに損傷が起きると核内で凝集するが、細胞質でSGの形成にはあまり関与しない。
家族性ALSにおける変異部位(点線)はprion-likedomainに集中している。FUSでは他に、RGG rich、PY-NLS 領域にも変異が見つかっている。ALS data browser (http://alsdb. org), and the ALS Online Genetics Database (http://alsod.iop.kcl.ac.uk/)より。
PY-NSL; proline-tyrosine nuclear localization signal
TDP-43の病態
TDP-43 proteinopathyはFTLDとALSを引き起こすが、その機序は、1)TDP-43の核内での減少に伴う機能低下、2)TDP-43が細胞質に凝集してリソソームやオートファジーなどの蛋白分解系に負担をかける、などの機序が推測されている。
TARDBPやFUSの遺伝子異常ではこれらの変異蛋白の機能低下(loss of function)によりALSを発症しやすく、これらの変異タンパクは細胞質に凝集しやすいと考えられる。他の遺伝子異常では、オートファジーやUPSの障害によりこれらの蛋白が細胞質に蓄積して蛋白合成系や分解系に負担をかける(gain of toxic function)ことにより、FTLDの発症リスクが高まる。
ALS/FTDの患者のTDP-43封入体の中には、TDP-43のCTF(C-terminal fragment)が多く含まれている。TDP-43が小胞体(ER)のカスパーゼよって切断された場合は、そのC末端側の破片であるCTF(C-terminal fragment)-25やCTF-35はprion-like domainを持つため重合しやすい状態と考えられている。これが重合する際にfull-lengthのTDP-43も引き込み封入体を形成すると思われる。小胞体では常に一定の割合で不良な蛋白が生成されており、カスパーゼはこのような蛋白を分解している。CTFはカスパーゼだけでなく、非リソソーム系プロテアーゼであるカルパインによっても生成されるが、なぜ封入体にCTFが多く含まれているのかはよくわかっていない。過剰なTDP-43の産生抑制やERストレスによるカスパーゼの活性化、細胞内カルシウム増加に伴うカルパインの活性化などの関与が推測されている。分裂して増殖する細胞においては、一定以上の不良蛋白が蓄積しても、カスパーゼの作用によりアポトーシスを起こして生まれ変わることができるが、神経細胞ではこのようなことができない。神経細胞のような長寿な細胞には徐々に不良蛋白が蓄積して、その生命活動が圧迫される可能性がある。
TDP-43以外の不均一核内リボ核酸蛋白(hnRNP)であるFUSやA1、A2/B1、F、Kなどの遺伝子異常もALS/FTDを引き起こすことが報告されている{27215579}。興味深いことに、ALSと同じ運動ニューロンの障害である家族性spinal muscular atrophy (SMA)の原因もhnRNP (G、M、Q、R)の遺伝子異常が原因であることがわかっている注1。
注1;SMA(常染色体劣性遺伝)の患者はSMN(survival of motor neuron)蛋白の mRNAのエクソン7の6位がCからTに転位している病気。この状態であるとpre-mRNAにおける異常なエクソン7の部位はhnRNP A1/2によってspliceされてしまい、この領域が欠損した不完全なSMNタンパク質しかできなくなる。Nusinersen Sodium (スピンラザ®) はエクソン7の下流にあるイントロンにASO(antisense oligonucleotide)を結合させこのspliceを抑制することにより、正常なSMNタンパク質を増加さる。SMNには核内低分子リボ核タンパク質(snRNP)などからなるspliceosomeの形成、リボ核タンパク質の生成、mRNAのtraffickingなどの働きがある{29872871}。核内でsnRNPによるpre-mRNAのスプライシングはカハール体(Cajal body)近傍のGemと呼ばれる部位で行われている。ALSでは核内のGemの消失が観察されており、TDP-43やFUSもGemにおけるspliceosomeに直接・間接的に関与していると推測されている。
FTLDとALSの患者の神経細胞内に認められるユビキチン陽性構造物はどちらも変性したTDP-43が主成分であることが判明したのは2006年で(1, 2)、この10年余りの間に多くの重要な研究が報告されている。ALSは上位運動ニューロンと下位運動ニューロンの変性をきたす疾患で、この同じ病態のスペクトラムにFTLDがあることから、前頭葉と側頭葉前半が運動系ニューロンと深く関わっていることがわかる。一方、側頭葉の後半、頭頂葉、後頭葉が知覚系ニューロンと関わっている。前頭葉、島回前部、側頭葉前部は私たちの行動における、動機付け、判断、計画、遂行、情動に関係しており、これらの領域の神経細胞やグリア細胞には何らかの脆弱性があり、TDP-43やFUSの蛋白異常が引き起こされると推測される。最近の研究は、FTLD/ALSの病態として細胞内vesicleの輸送、異常な蛋白の処理、リソソーム/オートファジーの機能障害が重要視されている。異なる遺伝子の異常がTDP-43という共通した蛋白の蓄積につながることから、FTLD/ALSの病態が解き明かされつつあり、近い将来にはこれまで不可能とも思われてきた治療法の開発機運が高まっている。
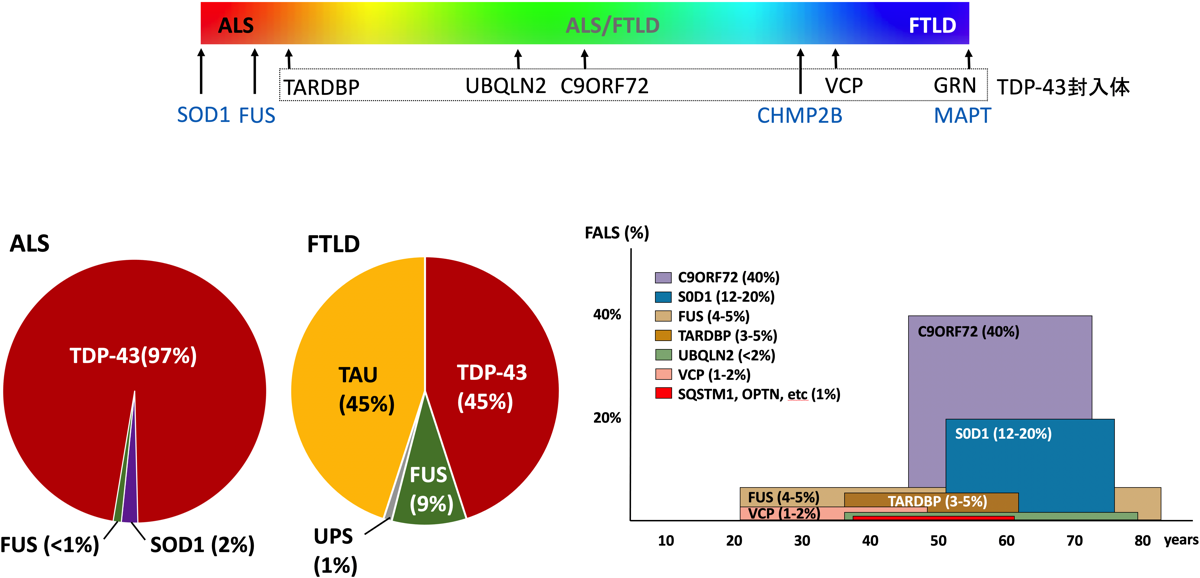
ALSとFTLD-TDPの原因には多くの共通点があるが、両者は全く同じではない。SOD1(superoxide dismutase)の異常はALSのみに認められる。TDP-43が関係する場合はALS、FTLDのどちらも発症するが、どちらが発症しやすいかは原因となる遺伝子の異常によって異る。
ALSの10〜15%は家族性(主に常染色体性優勢遺伝)で頻度の高い順にC9orf72、SOD1、FUS、TARDBP、UBQLN2、VCPなどの遺伝子異常が知られている。一方、FTLDで遺伝子異常が見つかる頻度はALSよりも高く30〜40%程度と言われているが、この中にはALSを発症しないタウ遺伝子であるMAPTやほとんどALSを発症しないGRNの遺伝子異常が含まれている。C9orf72、GRN 、TARDBP、VCP、UBQLN2の異常はTDP-43封入体が出現するので、これらはFTLD-TDPと分類される。孤発例も含めFTLDを変性タンパクで分類すると、TDP-43とtauが多くそれぞれ45%で、残りの9%がFUSになる。ALSではほとんど(97%)がTDP-43が関係しており、SODが2%、FUSは1%未満になる(図)。また、FTLDの15%にALS、ALSの15%にFTLDが合併すると言われている。FTLD-tauは3Rのものがピック病、4Rのものが、CBD、PSP、AGDとなる。
遺伝子異常とFTLD、ALSの発症頻度{23931993)}
以前にFTLD-Uと分類されていた大半が蛋白はTDP-43であることが判明し、TDP-43陰性のものをFTLD-Uと分類している時期があったが、後になってそのほとんどがFUSであることが判明している。現時点でFTLD-Uは、封入体のユビキチンが陽性でTDP-43やFUSが陰性のものを言うが、これに該当するものとしてCHMP2Bがある。
FTLDのタイプと遺伝子異常、封入体、ALS合併との関係を下表まとめた。FTLD-TDPの異常で多いのはC9ORF72とPGRNでTDP-43の遺伝子であるTARDBPの頻度は少ないことがわかる。この表で興味深いのは、CHMP2Bの変異がTDPやFUSの凝集を起こさない(下表参照)。
封入体と遺伝子異常 {27166223}
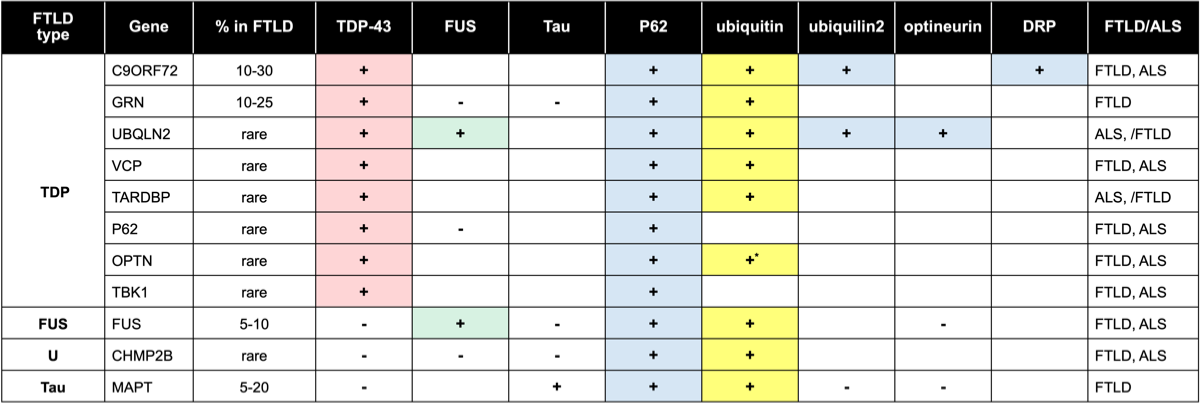
FTLD, ALS; FTLD or FTLD+ALS or ALS. ALS, /FTLD; ALS or ALS + FTLD
*文献{27552911}より
FTLD/ALSの発症に関係する蛋白とその機能
古くなった蛋白や合成時にできた不良品はユビキチンによってラベル付けされた後に処理される。これには、CASA、UPS、CMA、lysosomeを含めたvesicleの輸送などが関係しているが、FTLD/ALSの発症にはこのような蛋白除去機構の異常が関係している。図の赤字は遺伝子異常が見つかっている蛋白。リボソームによって生成された蛋白にはシャペロンが付着して正しい立体構造が取れるようにしている。異常な蛋白はカスパーゼで分解されるか、ERADを介してUPSで分解されることになる。TDP-43がカスパーゼで分解されると、凝集しやすいCTFが作られ細胞質に凝集塊を形成し、これが正常のTDP-43も巻き込んでしまう可能性がある。HSP70は細胞質の異常な蛋白を認識してubiqullin2と共にUPSに導き分解する。HSC70は凝集しかけた蛋白に結合してlysosomeで分解する(CMA)。凝集した蛋白はユビキチン化され、optineurinやp62が結合してphagophoreのLC3と結合してCASAで処理される。不必要になった細胞膜上のレセプター(グルタミン酸受容体など)にはユビキチンが結合してendocytosisを引き起こす。取り込まれたvesicleはearly endosomeと融合し、MVBに運ばれ選別される。MVBではESCRTタンパク群が作用して、vesicleを形成し蛋白の種分けを行う。CASAからautophagosomeに変化したvesicleはMVBと融合してamphisomeを形成し、やがてlysosomeと融合して蛋白は分解される。
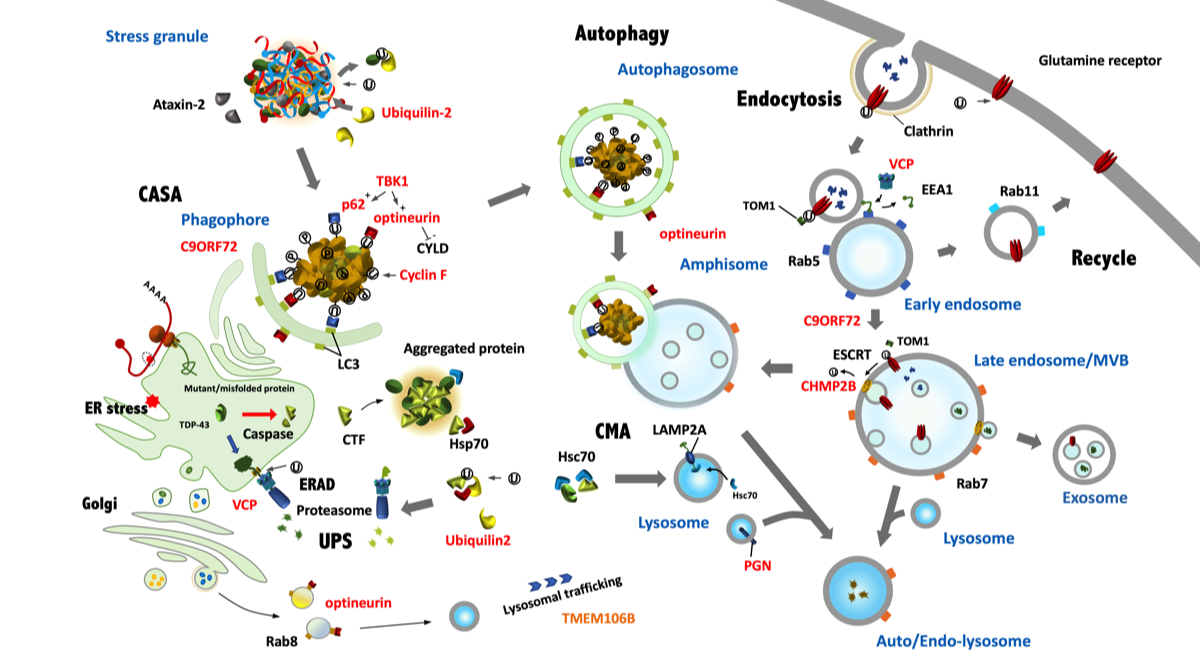
赤字は遺伝子異常が見つかっている蛋白。リボソームによって生成された蛋白にはシャペロンが付着して正しい立体構造が取れるようにしている。異常な蛋白はカスパーゼで分解されるか、ERADを介してUPSで分解されることになる。TDP-43がカスパーゼで分解されると、凝集しやすいCTFが作られ細胞質に凝集塊を形成し、これが正常のTDP-43も巻き込んでしまう可能性がある。HSP70は細胞質の異常な蛋白を認識してubiqullin2と共にUPSに導き分解する。HSC70は凝集しかけた蛋白に結合してlysosomeで分解する(CMA)。凝集した蛋白はユビキチン化され、optineurinやp62が結合してphagophoreのLC3と結合してCASAで処理される。不必要になった細胞膜上のレセプター(グルタミン酸受容体など)にはユビキチンが結合してendocytosisを引き起こす。取り込まれたvesicleはearly endosomeと融合し、MVBに運ばれ選別される。MVBではESCRTタンパク群が作用して、vesicleを形成し蛋白の種分けを行う。CASAからautophagosomeに変化したvesicleはMVBと融合してamphisomeを形成し、やがてlysosomeと融合して蛋白は分解される。
☞ HSP70 ファミリーである HSC70 はストレス顆粒内の異常な蛋白に結合したのち、lysosomeのLAMP2A (lysosome-associated membrane protein 2A)を介 して、lysosome内に導かれるCMAに関与している。 HSC70 は KFERQ モチーフがあると好んで CMA に誘 導しするが、タウ、α - シヌクレイン、TDP-43 はい ずれも CMA の対象になっている.。
☞ Rabは細胞のvesicle輸送に関係しているす。GTPが結合すると活性化されvesicleの膜に結合して輸送を促進するが、GDPと結合すると不活性となり細胞質に遊離する。グアニンヌクレオチド交換因子(GEF)はRabに結合したGDPを取り除きGTPに置き換えるが、DENNドメインを含む蛋白質はGEFの働きを助ける。
C9ORF72遺伝子
C9ORF72 (chromosome 9 open reading frame 72)の遺伝子異常はALSやFTLDを引き起こすが、この場合も病理でmotor neuronにTDP-43封入体が認められている。FTLD全体の10〜30%に関与しており、原因としては最も頻度が高いと考えられている。C9ORF72 遺伝子には6塩基反復配列であるGGGGCC(G; グアニン、C; シトシン)の繰り返しがありるが、通常この繰り返し数は20〜25までとされている。しかしながらALSやFTDの患者では、この繰り返しが異常に伸長しており、数百から数千にも及ぶ。C9ORF72 の異常は家族性ALSのみならず孤発性ALSでも見かけ、白人ではALS/FTDの孤発例で最も多く見つかる。半数近くはパーキンソニズムを伴い、DAT scanでも取り込みの減少が認められている{22807188}。日本では紀伊半島のALS症例にこの異常が見つかっている。C9ORF72 遺伝子異常では通常のALSやFTDの病理所見に加えて、小脳や海馬、前頭側頭新皮質の神経細胞にTDP-43陰性、ユビキチン陽性の封入体(p62/SQSTM1陽性)が認められるのが特徴で、これにはdipeptide repeat proteins (DPRs)が多く含めれている。GGGGCCの過剰な繰り返しは、C9ORF72蛋白合成のハプロ不全(片方の遺伝子の異常で蛋白量が減少する)を引き起こす。C9ORF72 の蛋白量の低下が、いかにしてTDP-43の細胞内蓄積を引き起こすかを解明することはALS/FTD の病態解明に役立つと思われる。
C9ORF72はDENN(differentially expressed in normal and neoplastic cells)ドメインを含む蛋白質で、Rabを活性化することによりvesicleの輸送を促進する。たとえば、Rab5、Rab7はendosomeからlysosomeに至るまでのvesicle輸送に関与している{29400714}。C9ORF72はRab1Aを介してULK1/2 (Unc-51 like autophagy activating kinase)をphagophoreに運搬してautophagyを起動させ、SMCR8、WDR41と共同でRab39Bを活性化してphagosome の形成を促進する{27103069}。C9ORF72の蛋白量の低下は細胞のautophagosomeの機能低下を引き起こして疾患を誘発すると考えられている{28824365}。
神経細胞ではシナプス形成の可塑性はグルタミン酸レセプターのendocytosisとその細胞内でのエンドソームでの分解によりる。C9ORF72に異常のあるニューロンではNR1(NMDA)とGLUR1(AMPA)レセプターが過剰に発現しており、グルタミン酸による細胞傷害を受けやすい状態になっている。不要なグルタミン酸レセプターはendocytosisによって取り込まれearly endosomeと融合しるが、これらもRabが関係しているため、C9ORF72の蛋白量の低下はautophagosomeによるグルタミン酸レセプターの数の調整に影響する(図) {29400714}。
C9ORF72 遺伝子 のGGGGCCリピート配列は本来翻訳されないはずであるが、リピート配列の伸長例では開始コドンを用いない翻訳(repeat-associated non-ATG-initiated translation, RANT) によって翻訳されてしまい、dipeptide repeat proteins (DPRs; グリシン-アラニン、-プロリン、-アルギニン)が大量に細胞内に蓄積されp62/SQSTM1陽性の凝集体として認められる{23381195}, {23415312}。このDPRsの蓄積はC9ORF72 遺伝子のリピート延長に特有のもので、少なくともこれらのペプチド合成はERにストレスを与えると思われるが、これがALS/FTD発症の機序に影響するかは不明。1) 6塩基反復配列であるGGGGCC(G; グアニン、C; シトシン)の繰り返し数が異常に多くなり、この長いリピート構造を持つRNAがTDP-43などのRNA結合タンパクを引きつけて凝集させる、2)本来翻訳されないGGGGCCリピート配列が翻訳されてしまい、dipeptide repeat proteins (DPRs; グリシン-アラニン、-プロリン、-アルギニン)が細胞質に蓄積して毒性を発揮する、ことが推測されている。
☞ Rab39bは神経細胞に特異的に発現しており、AMPAレセプターのサブユニットであるGluA2のシナプスでの輸送に関与している。Rab39bの遺伝子異常はレビー小体病を引き起こしる。Rab7、Rab2B、Rab11の変異はCarcot-Marie-Tooth diseaseを引き起こす。
GRN遺伝子
GRN(granulin)遺伝子の異常はALSではなくFTLDを引き起こす。その支配下のプログラニュリン(progranulin)は糖蛋白で、神経を含め色々な細胞から分泌されて、細胞の成長、腫瘍形成、創傷治癒、炎症反応、インスリン抵抗性などの反応を引き起こすと言われているが詳しいことはわかっていない。脳では神経突起の成長やミクログリアによる炎症応答の制御に関与している。プログラニュリンの産生低下は関節リュウマチの原因と言われている。プログラニュリンの産生はGRN遺伝子の支配下にあるが、対立遺伝子の一方に障害がある(haploinsufficiency)があるとTDP-43封入体のあるFTLDを発症する(常染色体優性遺伝)。さらに遺伝子異常がホモの場合(homozygous mutation)には神経性セロイドリポフスチン症(NCL; neuronal ceroid lipofuscinosis)を発症する(常染色体劣性遺伝)。FTLD全体の10〜25%を占め、FTLD-TDPの原因としてはC9ORF72に次いで2番目に多い。
ヒトのプログラニュリンはグラニュリン(granulin)が7.5個繋がった構造をしており、sortilinによってlysosomeに輸送されてカテプシン-Lで切断されるとそれぞれ作用が異なる7種類のグラニュリン(granulin-G, -F, -B, -A, -C, -D, -E)とparagranulinが作られる。グラニュリンは細胞外ではサイトカインのように働き、プログラニュリンの抗炎症とは逆の作用をするが、その生理作用はほとんどわかっていない。細胞内ではlysosomeにとどまり、lysosomeの作用を増強すると推測されている。グラニュリンは12個のシステインをもち、いわゆるcysteine-rich mini-proteinに属し、このグループにはオキシトシン、ソマトスタチン、インターロイキン-8、上皮成長因子、defensinなどがあり、グラニュリンにも似たような作用が推測されている。
プログラニュリンが低下するとTFEB遺伝子が活性化されてTFEB(transcription factor EB)蛋白合成が増加するが、これはlysosome形成の遺伝子転写全体を促進する。結果として、lysosomeの数が増加、lysosomeの酵素が増加、autophagyの増加、lysosomeのexocytosisが増加する。プログラニュリンの低下は、ミクログリアのphagocytosisを助長し、pro-inflammatory cytokinesが増加する。結果としてミクログリアによるシナプスの剪定作用が過剰になる可能性がある。一方でプログラニュリンには、lysosome酵素のシャペロンとしての作用や酸性化の作用があり、プログラニュリンの低下は、lysosomeのグラニュリンの減少とあいまってlysosomeの機能低下を招く。Lysosomeの機能不全は神経細胞の老化を促進し、TDP-43が細胞質内に蓄積しやすくなる可能性がある。TDP-43以外にもリン酸化タウ蛋白やα-シヌクレイン蛋白も蓄積していることが観察されており、アルツハイマー病におけるタウ蛋白の蓄積やパーキンソン病との関連も指摘されている。Lysosomeにおいて、プログラニュリンとグラニュリンEはカテプシンEを活性化するが、この機能の低下はTDP-43の細胞内沈着と関係している。
プログラニュリンの低下は神経性セロイドリポフスチン症やゴーシェ病を発症させるが、興味深いことにプログラニュリンはHSP70と共にlysosomeでβグルコシダーゼとカテプシンDのシャペロンとして働いている{27789271}{28453791}。神経性セロイドリポフスチン症はlysosomeの酵素であるカテプシンDの変異でも出現する(type 10)。また、プロサポシン(prosaposin)はカテプシンDで4種類のサポシンになるが、サポシンの低下はゴーシェなどのスフィンゴリピドーシスの原因となる。LysosomeのカテプシンDの低下はALS/FTLD-TDPやアルツハイマー病の発症リスクを高めると言われている。
☞ プログラニュリンのA、C、FユニットはTNFレセプターに結合するが、A、C、Fユニットから人工的に作られたAtsttrinはTNF-αのsignalingを抑制することにより、リウマチやクローン病の治療に役立つことが期待されている{29258611}。
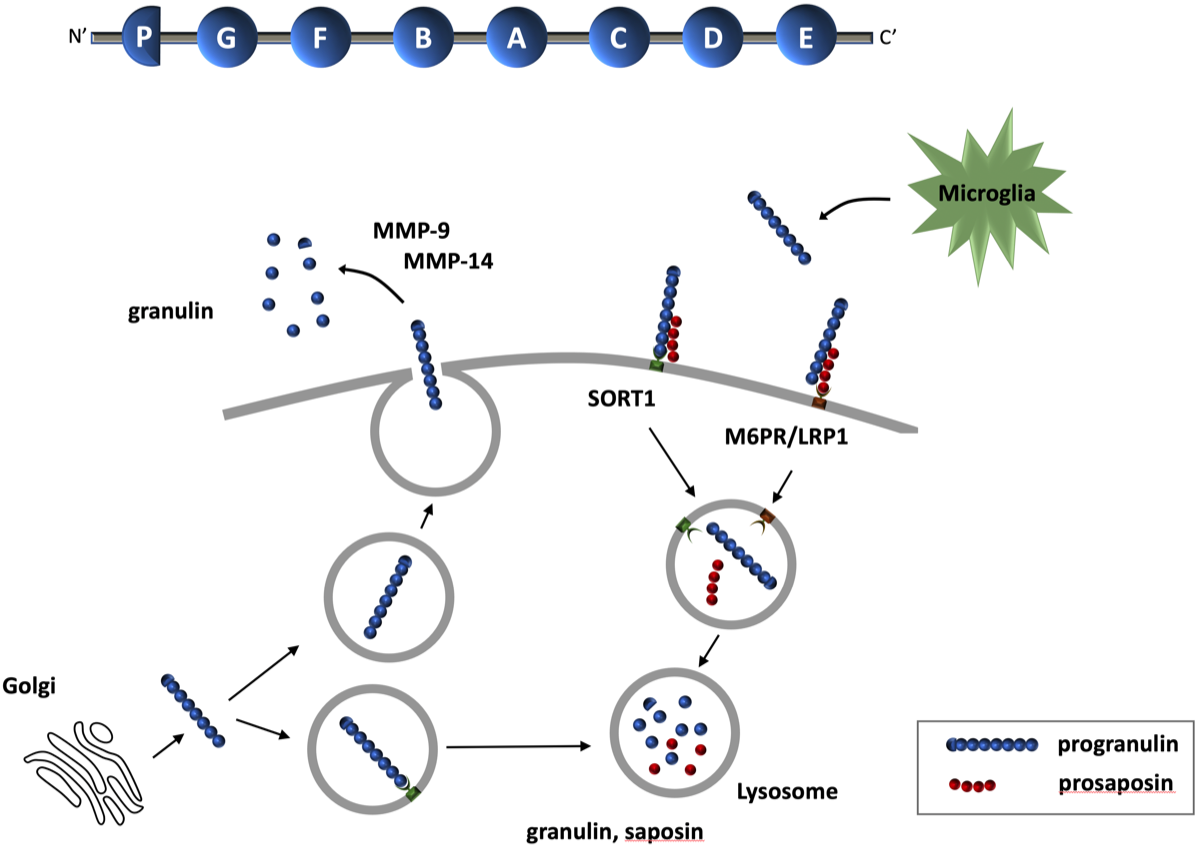
プログラニュリンはsortilin(SORT1)と結合してlysosomeに輸送される。ミクログリアからも分泌され、細胞表 面でsortilinと結合、あるいはprosaposinと結合してLRP1(low-density lipoprotein receptor-related protein 1)を 介してlysosomeに運ばれる。プログラニュリンはlysosomeで7種類(G,F,B,A,C,D,E)のグラニュリンと パラグラニュリンになり、プロサポシンは4種類のサポシン(A,B,C,D)になる.プログラニュリンは細胞外 に分泌されて、matrix metalloproteinase(MMP)-9やMMP-14によってグラニュリンに分解される.プログラニュ リンのA,C,FはTNFレセプターに親和性を持つ。
VPC遺伝子
Valosin-containing protein (VCP)またはp97は、AAA (ATPase associated with diverse cellular activities) に属する蛋白で、ユビキチン化された蛋白の立体構造をほぐす(unfolding)ことで、小胞体やミトコンドリアの蛋白の品質管理、DNA修復、細胞周期の調整などを担っている。MVB(multivesicular body)におけるESCRT (endosomal sorting complexes required for transport) の働きにも関係して、樹状突起の剪定に影響を与えている。
小胞体で正しい立体構造が取れなかった蛋白がユビキチン化されると、VCPは小胞体膜のSec61チャンネル(translocon)に結合して、この不良蛋白を小胞体から細胞質に引き出し、プロテアソーム(26S)に導いて分解する。この一連の過程は小胞体関連分解(ER-associated degradation; ERAD)と呼ばれている。ミトコンドリアの不良蛋白に対してもVCPによって同様の処理が行われている (mitochondria-associated degradation; MAD)。VCPは2つのATPaseドメインを持ち、N 末端側で基質と結合する。VCPは6 個のサブユニットがリング状に集まり、これが上下の2重リング構造を形成している。それそれのユニットに1個のAAA型ATPaseドメインを配置している。リングの中を蛋白が通過する際に上(D1)と下(D2)のそれそれのサブユニットがATP依存性に別々の動きをすることにより蛋白を通過させ、立体構造をほぐす。ほぐれた蛋白はそのまま26Sプロテアソームによって分解される(UPS)。同様に細胞質においてもユビキチン化された不良蛋白にVCPが結合してUPSに導く。
DNAが2本鎖とも損傷を受けている(DSBs; DNA double-strand breaks)場合の修復機構の1つに非相同末端結合がある。切断されたDNAの断端をKu70/80 heterodimerが認識して修復を促しる。DNAが修復されるとリング状のKuはDNAから離れられなくなりるが、VCPはユビキチン化されたKu80に結合して、Ku70/80を引き離す{27716483}。
NSF(N-ethylmaleimide-sensitive factor)とVPS4(vacuolar protein sorting-associated protein4)はVCPと同じAAAに属する蛋白でそれぞれの構造も極めて類似している。
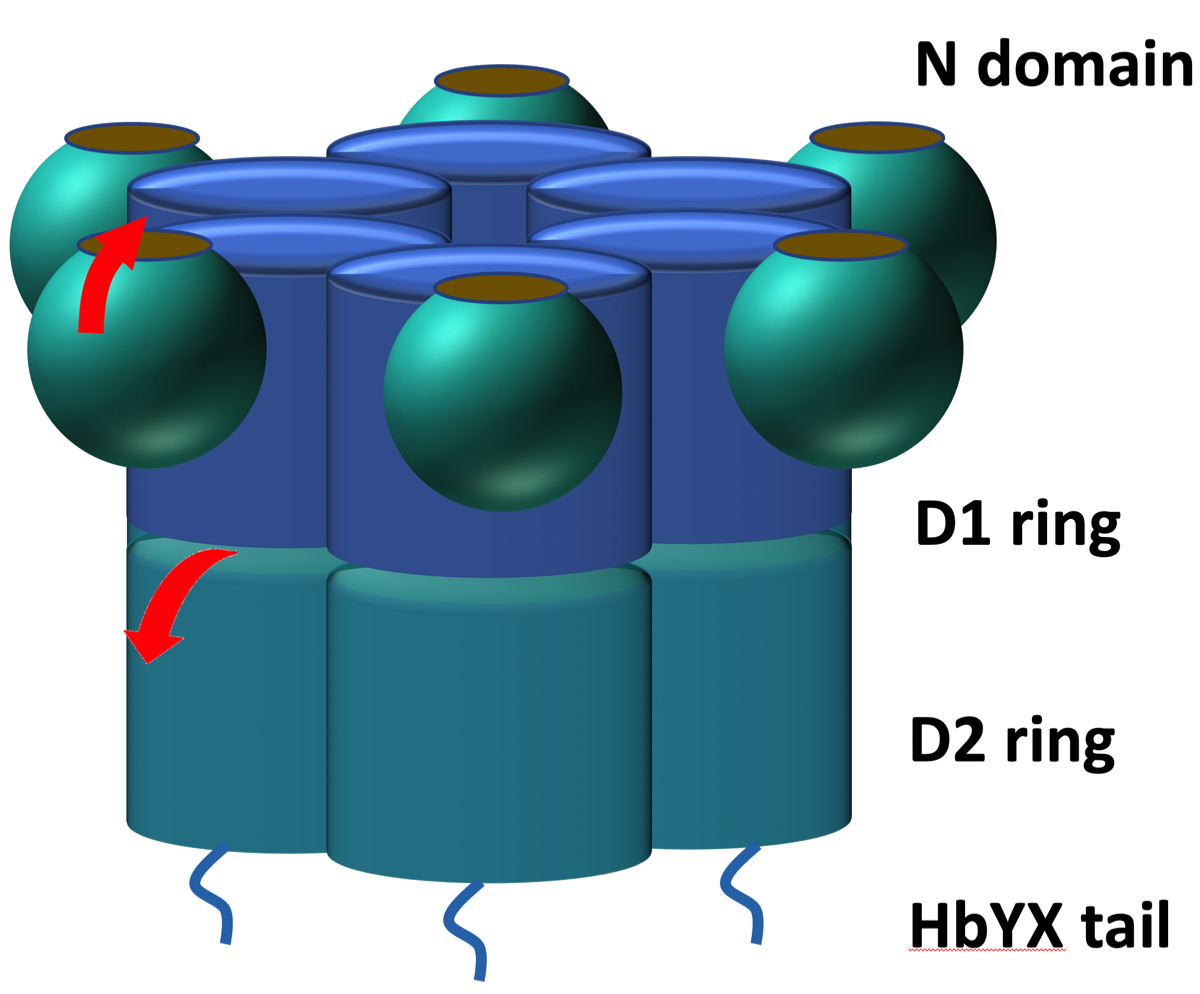
D2のATPが加水分解されると、赤矢印の方向に10 〜 15°回転 する。その後D1のATPが加水分解されると、赤矢印のようにN ドメインが持ち上がるように動く。
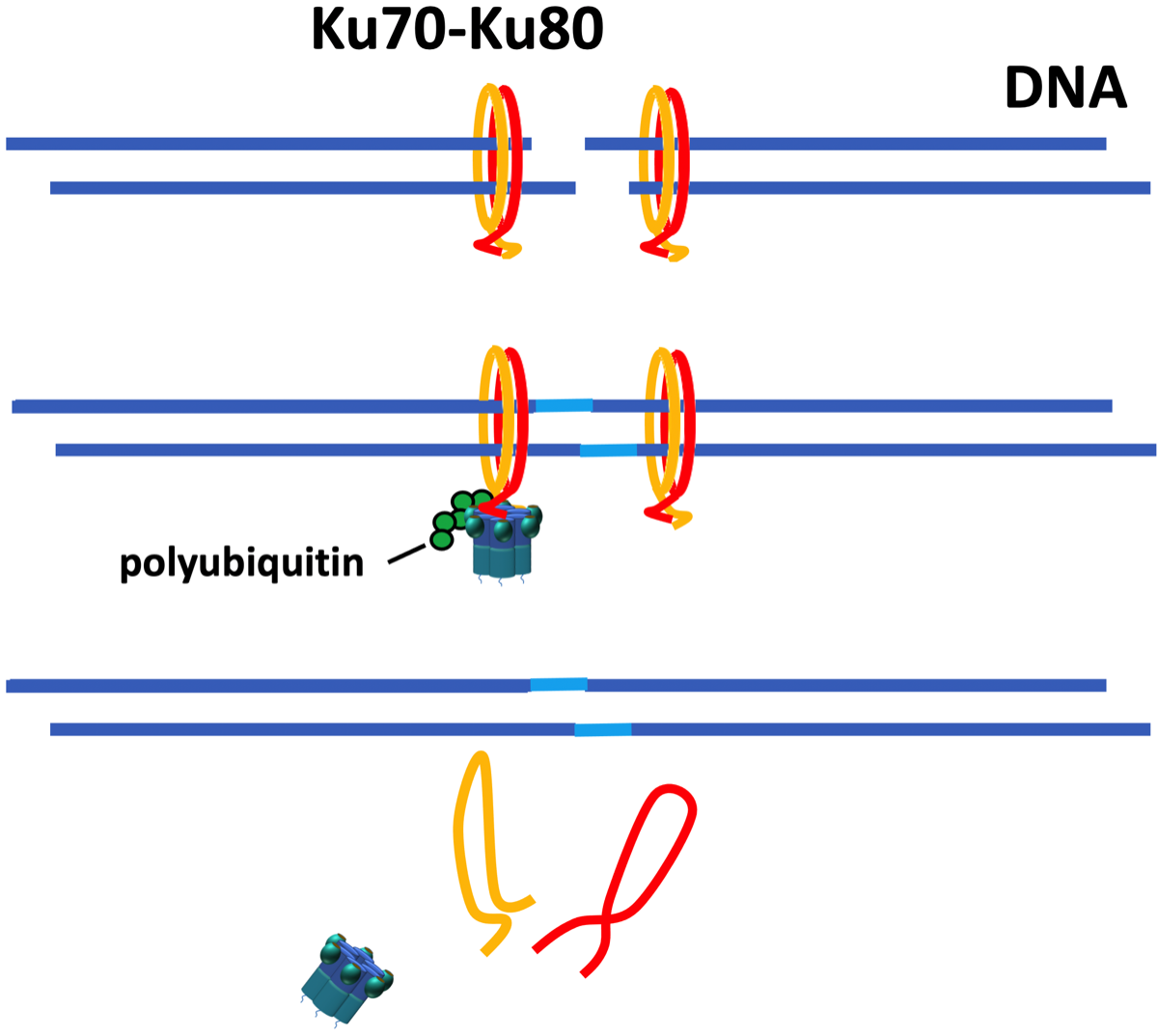
DNAが2本鎖とも損傷を受けると、リング状のKu70-Ku80ヘテロ二量体が損傷部位を発見し、DNA-PKcs、XLF、Artemis、DNA ligase IVなどにより一連のDNA修復作業が始まる。DNAが修復されると、そのままではKu70-Ku80ヘテロ二量体は外れないため、ユビキチン化後にVCPが作用してKuを解放する。
NSFとVCPは膜融合時のSNARE蛋白の絡みを解放して再利用する働きを担っている。NSFは融合後のcis SNARE complexを解放するが、VCPも同様にSNAREの解放作用があると考えられている。一方、VSP4はCHNP2Bに関連するESCRT-IIIサブユニットを膜から解放する際に働く{14514884}}。EEA1(early endosomal autoantigen 1 )によるvesicleの融合にVCPがNSFと同じような働きをしている可能性が示唆されている{21556036}。
FTLD/ALSを引き起こすVCPの変異のほとんどは基質と結合するN末端側に起きている。その遺伝子異常は、骨パジェット病(Paget's disease of bone; PDB)および前頭側頭型痴呆をともなう封入体ミオパチー(inclusion body myopathy with Paget's disease of bone and frontotemporal dementia; IBMPFD)を引き起こすことが知られている。Proximo-distal inclusion body myopathy (IBM)、骨パジェット、FTLD、ALSのどの症状をどの程度示すかは、同一家系内であっても様々である。もっとも高頻度なのは封入体ミオパチー(封入体筋炎も含む)で約90%に認められ、30%は筋炎のみの症例となっている。骨パジェットは50%、FTLDは30%でALSは稀である。Charcot-Marie-Tooth (type2)、統合失調症への関与も報告されている。この疾患で興味深いのは、TDP-43封入体は筋細胞では細胞質であるのに対し神経細胞では核内であることで、その理由はわかっていない。
プログラニュリンの低下は神経性セロイドリポフスチン症やゴーシェ病を発症させるが、興味深いことにプログラニュリンはHSP70と共にlysosomeでβグルコシダーゼとカテプシンDのシャペロンとして働いている{27789271}{28453791}。神経性セロイドリポフスチン症はlysosomeの酵素であるカテプシンDの変異でも出現する(type 10)。また、プロサポシン(prosaposin)はカテプシンDで4種類のサポシンになるが、サポシンの低下はゴーシェなどのスフィンゴリピドーシスの原因となる。LysosomeのカテプシンDの低下はALS/FTLD-TDPやアルツハイマー病の発症リスクを高めると言われている。
UBQLN2遺伝子
UBQLN2遺伝子はX染色体にあるため、女性の発症リスクは低くなっている。蛋白が新生される際にシャペロンとしてHSP70が結合するが、出来上がった蛋白に異常があるとユビキチン化されるため、これにUBQLN2 (ubiquilin2)蛋白が結合してHSP70と複合体を形成する。この複合体はプロテアソームに導かれて、UPSで分解されることになる{27477512}。UBQLN2陽性封入体はTDP-43やFUS、SOD1によるALS/FTLD以外にアルツハイマー病、レビー小体病などでも認められている。UBQLN2はストレス顆粒中のユビキチン化された蛋白にも結合してこれを除去していると考えられている{29526694}。
☞ HSP70ファミリーであるHSC70はストレス顆粒内の異常な蛋白に結合したのち、lysosomeのLAMP2A(lysosome-associated membrane protein 2A)を介してlysosome内に導かれるシャペロン介在オートファジー (chaperone mediated autophagy; CMA)に関与している。HSC70はKFERQモチーフがあると好んでCMAに誘導するが、タウ、α-シヌクレイン、TDP43はいずれもCMAの対象になっている。
OPT遺伝子
OPTは遺伝性の原発性開放隅角緑内障(POAG, primary open-angle glaucoma)に関係するoptineurin(OPTN)をコードする遺伝子として発見されたもので、その名称はoptic neuropathy-inducingに由来する。 POAGはOPTNの他TBK-1(TANK-binding kinase 1)の遺伝子異常でも発症することが知られている。TBK-1はOPTN やp62をリン酸化してLC3とユビキチンの結合を強める。OPTNはALS以外に、ページェット病、クローン病にも関係している可能性がある。変異のないOPTNは、ALSやFTLD以外にADやパーキンソン病の神経細胞の封入体でも見つかっているが、変異OPTN(OPT遺伝子異常)がFTLDの原因になるかどうかは議論の余地がある{21074902}。
Nuclear factor kappa B (NF-𝛋B)シグナルは炎症やアポトーシスを引き起こしるが、OPTNはNF-𝛋B を活性化するNEMO(NF-𝛋B essential modulator)と構造がよく似ている。OPTNはTAX1に結合してNEMOと競合し炎症を抑制する{28456633}。OPTNの遺伝子異常のうち、ALSを引き起こすタイプでは抗炎症作用の増強、POAGを引き起こす遺伝子変異では低下(炎症亢進)が観察されている{29875767}。TDP-43やFUSの変異は神経細胞質に凝集するが、変異OPTNは神経細胞に凝集塊は形成しない。
OPTNはmyosin VI とともにexocytosisの際の小胞輸送に関与している。ゴルジ体のトランス面で、小胞表面にあるRab8に結合して、小胞(secretory vesicle)を細胞表面に輸送して細胞膜と融合させる{22801549}。同様にhuntingtin蛋白と結合してリソソームの輸送にも関与している。また、OPTNはp62と同様にTBK-1で修飾を受けたのち、ポリユビキチン化蛋白に結合してchaperone-assisted selective autophagy(CASA) に関与している。
CHMP2B遺伝子
CHMP2B(charged multivesicular body protein 2B/chromatin-modifying protein 2B)はESCRT-III(endosomal-sorting complex required for transport)に属する蛋白で、主にlate endosomeにおけるvesicle形成に関与している。ESCRTは0、I、IIの順にendosome膜に作用して、内側(intraluminal vesicle)あるいは外側に小胞(exosome)を出芽させるが、ESCRT-IIIは小胞の切り離しに作用し、その後VSP4によって膜から解放される{17450176}。興味深いことに、CHMP2BはCHMP4BとALIXと共同でdendritic spineの出芽にも関係しており、その異常はFTLDの発症の関係が示唆されている{26910595}。
Dendritic spineの表面にあるNMDAやAMPA receptorは常に破壊されたりリサイクルされたりしている。AMPA receptor数の増加と減少はそれぞれLTPやLTDの際に認められ、この変化はシナプスの可塑性や学習効果に関係している。Dendritic spineが活動するとその活動量に応じてlysosomeがspine内に輸送される{28724526}。過剰なAMPA receptorの発現はALSやFTLDの原因になると言われているが、CHMP2BはMVBやlysosomeにおけるdendriteのreceptor数を調整に関与している。Dendritic spineはマッシュルームのような形状をしているが、このくびれの形成にはCHMP2B を含めたESCRTが関与し、dendrite膜上のレセプターなどが拡散するのを防いでいる {28811236}。CHMP2B遺伝子異常による神経細胞にはTDP-43やFUSの凝集が認められないため、病理ではFTLD-Uに分類される。
シナプスの可塑性とAMPAレセプター
AMPAとNMDAレセプターは神経細胞のシナプスにおいて共存している。どちらのレセプターもグルタミン酸が結合するとNa+やCa2+などの陽イオンが流入する。AMPAレセプターは4量体で形成されているが、GluA1、2、3、4の4種類のユニットの中で、成人ではほとんどがGluA1-2とGluA2-3の組み合わせでできている。GluA2ユニットが含まれるとAMPAレセプターはCa2+を透過させないことから、ほとんどのAMPAレセプターはCa2+を透過しない。AMPAはその数に依存して活動電位に対する反応が大きくなるが、これはGluA1のリン酸化と脱リン酸化によって調節されている。一方、NMDAはMgでブロックされておりグルタミン酸が結合してもチャンネルは開かない。このような状態ではAMPAだけがシナプス後電位を引き起こしている(EPSP; excitatory postsynaptic potential)。
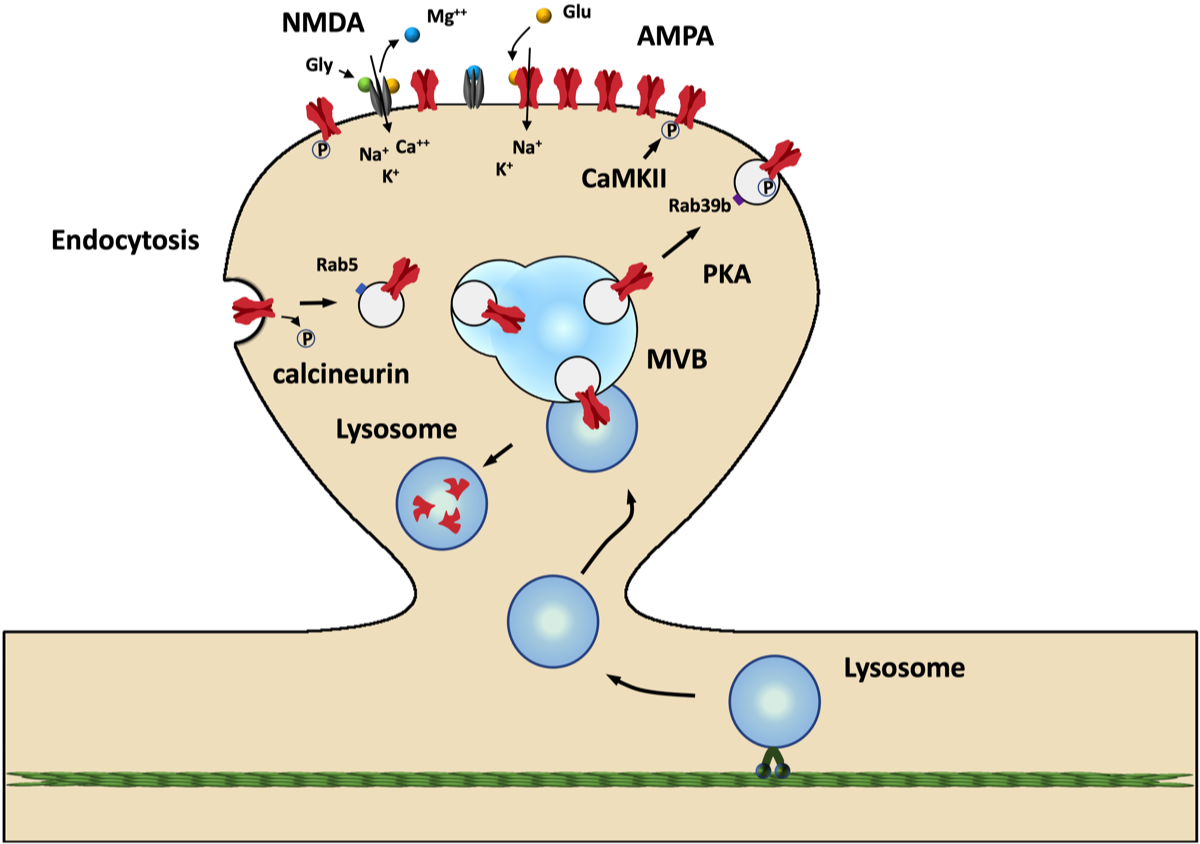
AMPAレセプターはシナプスの活動に応じて数が調節されているが、endocytosisとlysosomeの輸送はこれに大きく関わっている。
EPSPが連続的に起こり強い脱分極が生じるとNMDAのMgが外れてNMDAもグルタミン酸に反応するようになる。この時の刺激が高周波でシナプスに流入するCa2+が多い時は、cyclic-AMP–dependent protein kinase (PKA)の活性化によりGluA1がリン酸化されて、そのシナプスにおけるAMPAレセプターの数が増加してシナプスの伝達効率が増加する(LTP; long term potentiation)。また、calcium/calmodulin dependent protein kinase II (CaMKII) やprotein kinase C (PKC)の活性化によりGluA1の別の部位がリン酸化されるとAMPAの反応が高まる。一方、低周波刺激の場合はシナプスに流入するCa2+が少ないため、calcineurinが刺激されAMPAはendocytosisによって取り込まれる(LTD; long term potentiation)。LTPが長期に及ぶと一時的に増加したAMPAの一部はNMDAに置きかわり安定化する{17418904}。Lysosomeは微小管に沿って樹状突起に積極的に運搬され{28630145}、シナプスにおけるAMPAの調節に関与している。FUSはAMPAレセプターのmRNAの安定化に関係しているという報告がある{25968143}。
Lysosomeの運搬障害はFTLDの原因になるが、これと関連してtransmembrane protein 106B (TMEM106B)が注目されている。Single-nucleotide polymorphisms (SNPs) の検討で、TMEM106BがFTLD-TDPの発症リスクと関係していることが明らかにされた{20154673}。TMEM106Bは膜1回貫通型の糖タンパク質でlate endosomeやlysosomeに存在し、lysosomeの樹状突起への輸送に関与している{24357581}{24357581}。
☞ポリグルタミン病の1つである脊髄小脳変性症2型(SCD; spinocerebellar degeneration, type2)は、ATXN2遺伝子のCAG(グルタミン酸のコドン)リピートが異常に多い常染色体優勢遺伝する疾患として知られている。ATXN2遺伝子から翻訳されるataxin-2蛋白のQ(グルタミン酸)の繰り返しは通常22前後であるが、これが35を超えるとSCD-2(主にプルキンエ細胞の障害)を発症する。興味深いことに、繰り返し数が27〜33の中間程度だとALSの発症リスクが高くなると言われている。病態はよくわかっていないが、本来、ataxin-2は細胞質内においてmRNAの3’UTRに結合してmRNAを安定化させる作用がある。ストレス顆粒形成において、ataxin-2がTDP-43を引き込むことことが推測されているが、これにはpoly-Qの数が影響している可能性がある。SMAと同様にASO (antisense oligonucleotide)を用いてataxin-2の産生を抑制することにより、ALSやSCD-2の発症を抑制する治療法が示唆されている{28405020}。TDP-43を過剰発現させるだけでマウスは急速にALSを発症して1ヶ月で死にるが、Ataxin-2遺伝子を欠損させたマウスでは病気の発症は遅れ、生存期間も1年以上に延長した。
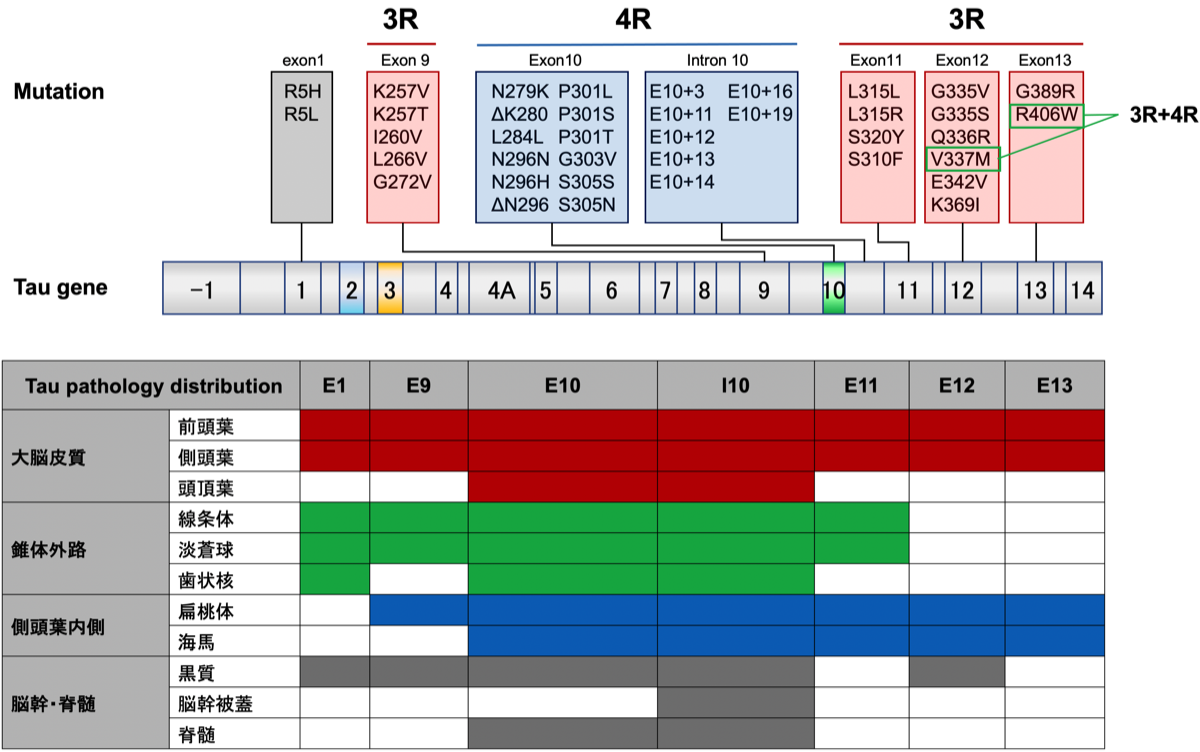
3Rタウオパチー(ピック病)は前頭葉と側頭葉に限局した障害を起こしやすい。これに対して4Rタウオパチーは基底核や小脳歯状核、側頭葉内側、黒質などの障害も出現しやすい。
FTDにパーキンソン症状を呈するFTD疾患
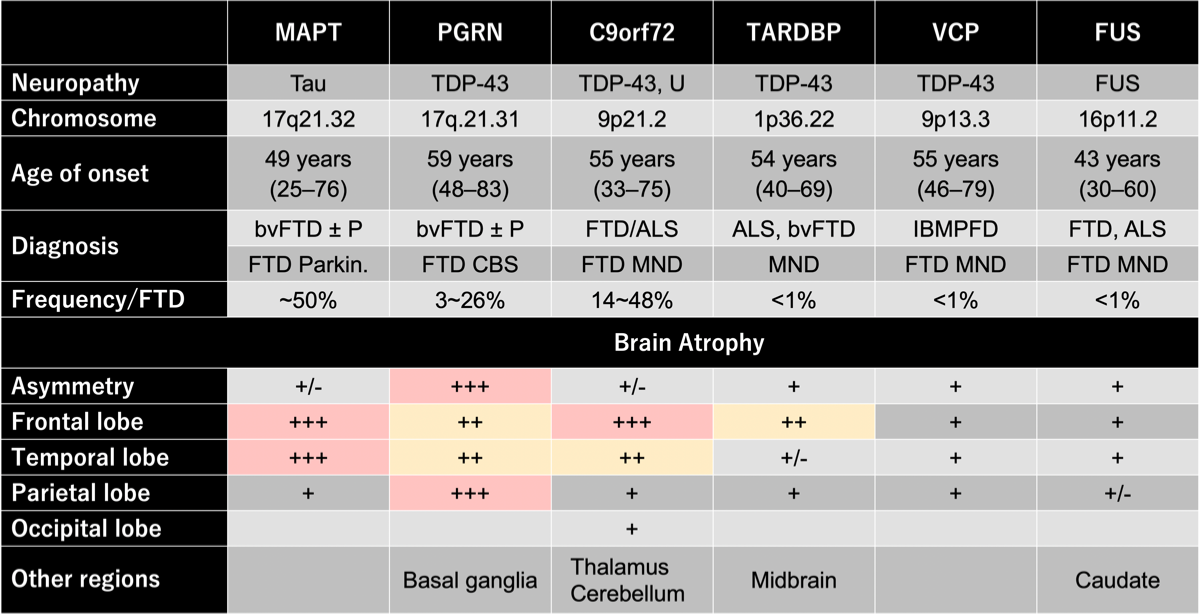
左右対称的な前頭側頭葉の萎縮が見られる場合にはMAPT、非対称的で頭頂葉にも萎縮が認められる場合にはPGRNを疑う。PGRNの症例は脳萎縮の進行が早いと言われている。萎縮が後頭葉や小脳にも認められる場合にはC9orf72を疑う。残りのTARDBP、VCP、FUSは頻度としては稀で、いずれも前頭葉、側頭葉、頭頂葉の萎縮が認められる。
側頭葉内側の萎縮があり、症状もアルツハイマー病に似ているがアルツハイマー病病理(ADNC)はなく、TDP-43封入体が神経細胞の細胞質に認められる。他、神経細胞の核や神経突起、オリゴデンドログリアやアストロサイトに異常にリン酸化されたTDP-43が蓄積している。このようなTDP-43の異常と脳萎縮の領域は一致して認められる。LATEの多くはADNCと合併していることが多い。純粋なLATEではADよりも進行はゆっくりであるが、進行すると寝たきりになる症例も存在する。ADNCと合併している場合は、純粋なADNCよりも進行は早い。まだ不明であるが、ADNCはLATE発症の危険因子の可能性がある。
LATEの病理所見は、80歳を過ぎた高齢者では20%以上の頻度で認められる。高齢者の海馬硬化症の多くはLATEが原因と思われるが、海馬硬化はTDP-43以外に虚血、てんかん、低血糖などでも認められる。
LATEは3ステージに分けられる。
ステージ1:扁桃体のみ (MMSE21±9)
ステージ2:+海馬、嗅内野 (MMSE=18±10)
ステージ3:中前頭回 (MMSE=14±10)
海馬硬化は重度のLATEに認められるが、40〜50%は片側性であり(進行すると両側性になってくる)、両側の海馬の萎縮が認められるFTLD-TDPとは異なる。LATEはA型FTLD-TDPに見られる特徴に類似していると指摘されているが、LATEは発症年齢が高く、認知症が主症状であり、病変ぶいの主座は大脳辺縁系である。症状は経験記憶の障害であるが、単語リストの遅延再生(海馬依存)が悪い割に、VFT(verbal fluency)=言語の流暢性(皮質依存)が保たれている。純粋のADNCと比較して、LATEが併存している症例では焦燥感・攻撃性のリスクが高まる。
LATEのMRIは、側頭葉内側の萎縮だけでなく、前頭葉下部、前部側頭葉、島皮質にも萎縮が認められる。
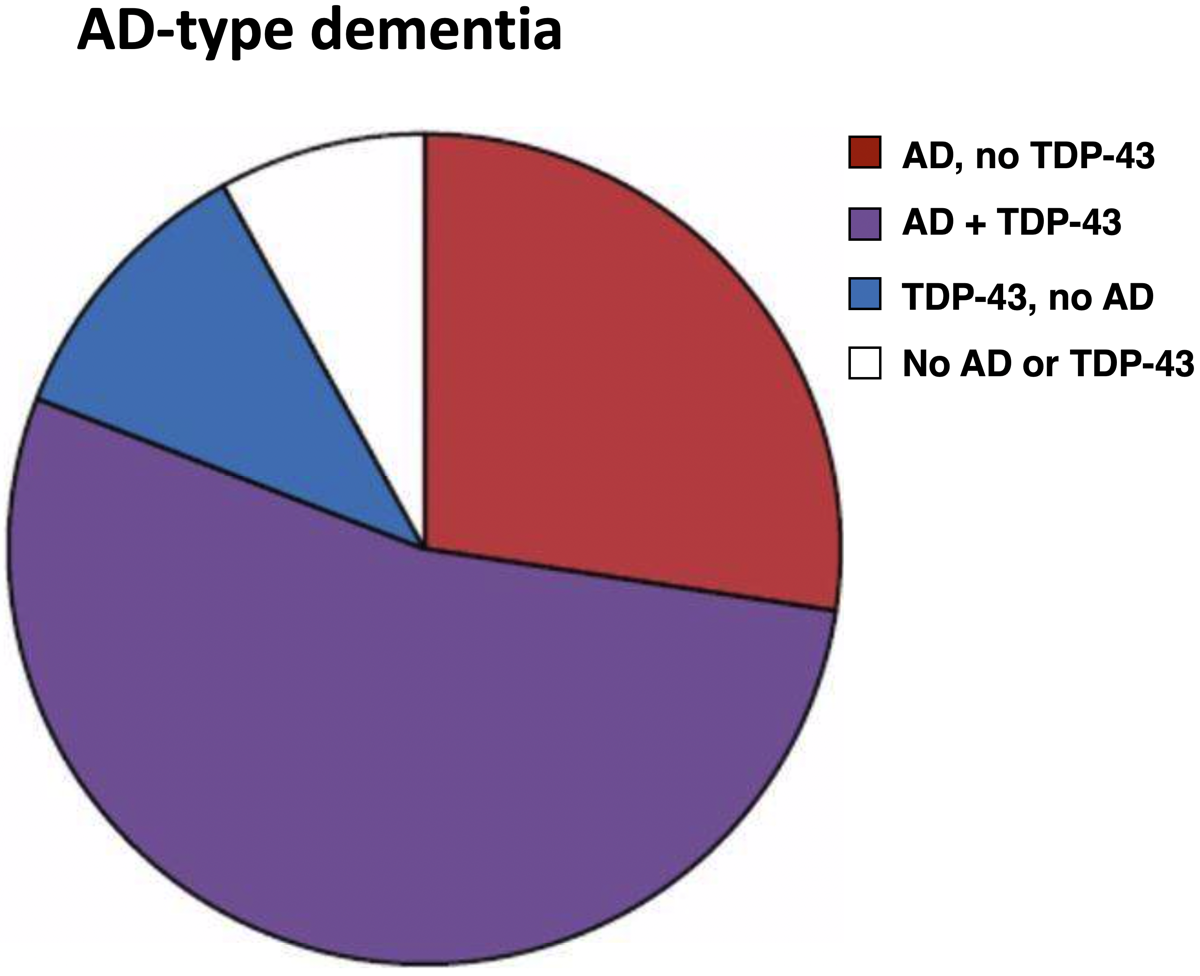
(James BD, et al. Brain, 2016) {27694152}
LATEの脳萎縮領域
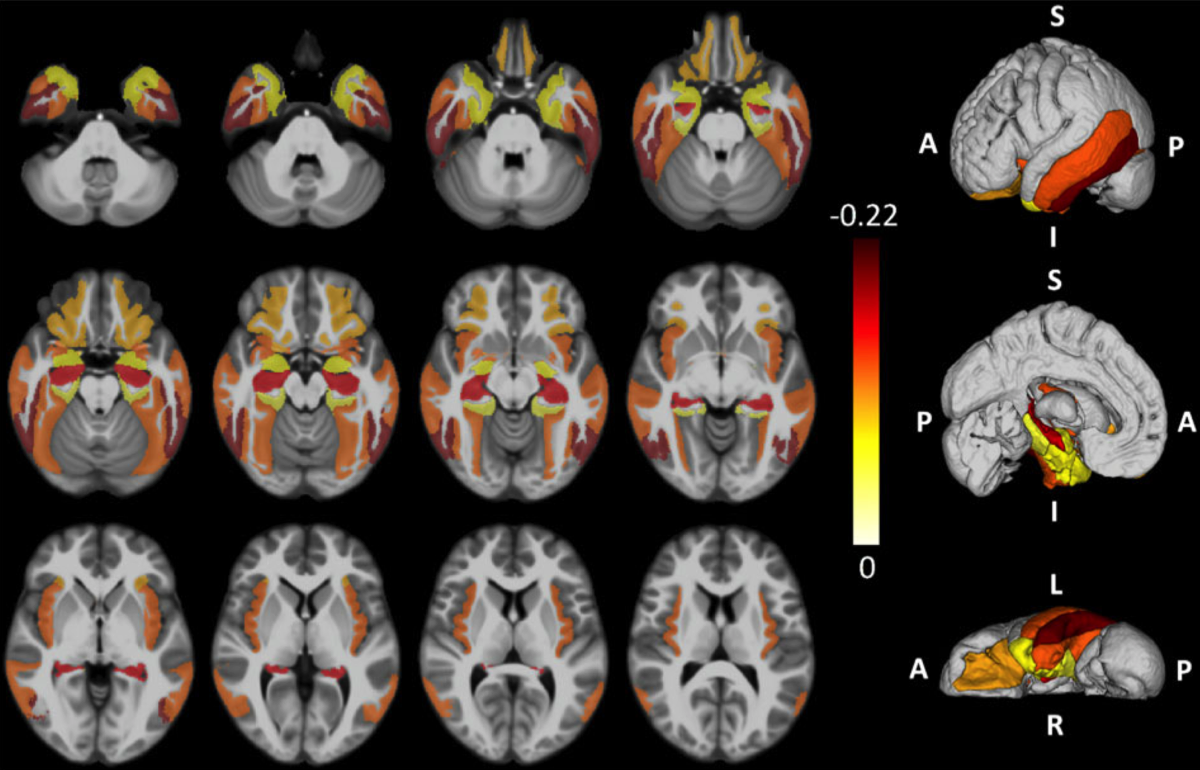
(Nelson PT. et al., Brain, 20119) {31039256}
LATEに関連する遺伝子として5つが指摘されている。17番染色体のgranulin (GRN)、7番染色体の transmembrane protein 106B (TMEM106B)、12番染色体のATP-binding cassette subfamily member 9 (ABCC9)、3番染色体のpotassium channel subfamily M regu-latory beta subunit 2 (KCNMB2)、19番染色体のapolipoprotein E (APOE) である。

LATEのまとめ
1. 80歳以上の症例で、海馬、特に前部の海馬の萎縮や扁桃体の萎縮が目立つ。
2. 後部帯状回の萎縮や代謝の低下が目立たない?
3.. 海馬硬化と診断されたことがある。
4. 高齢発症の側頭葉てんかん、一過性てんかん性健忘(TEA)
5. 経過観察中に易怒性や攻撃性が出現してくる(性格の変化)
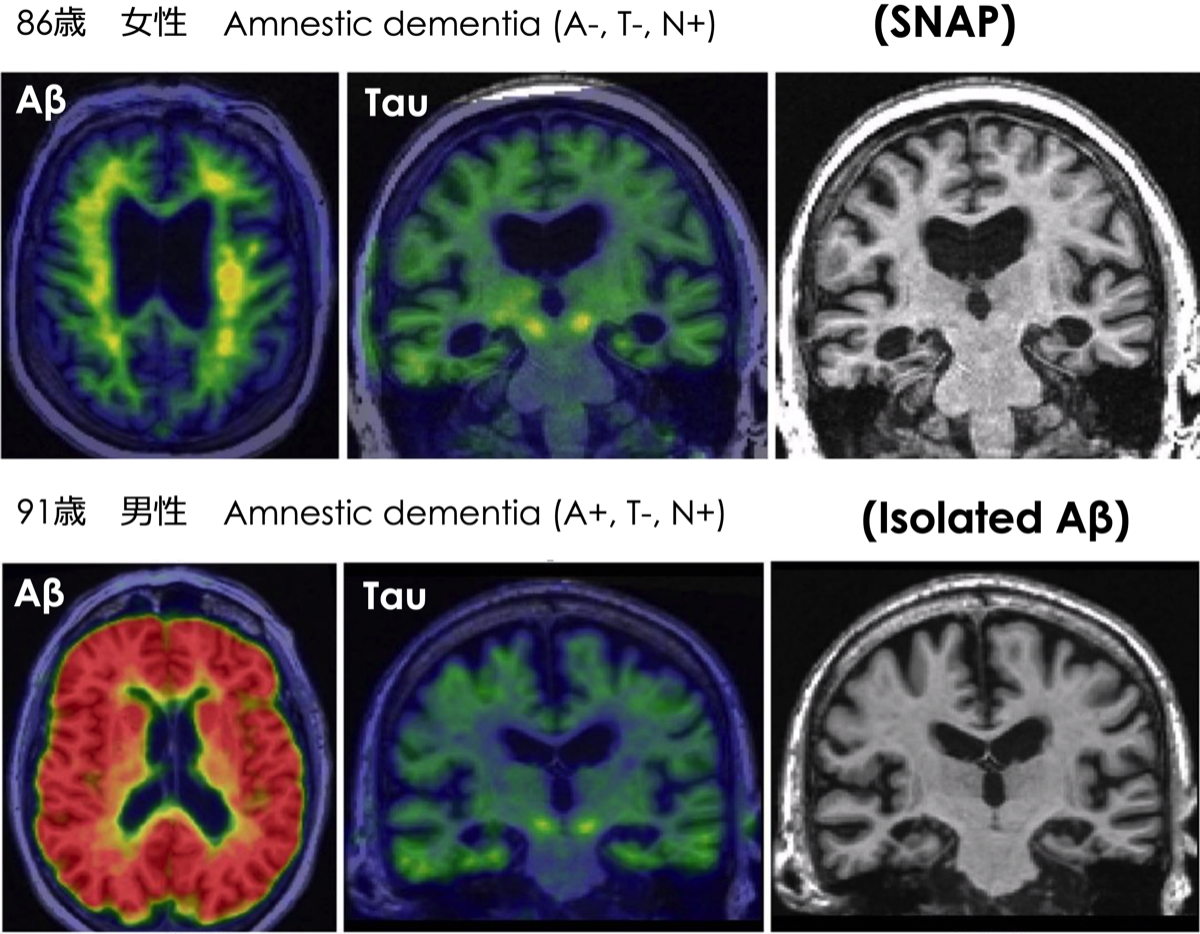
(A) 86歳の女性で、「Probable Alzheimer's disease」と診断された。しかし、アミロイドPETは陰性、タウPET検査も陰性、MRIでは両側の側頭葉内側に顕著な萎縮が見られた。この組み合わせは「A-T-N+」とされ、生前は「非アルツハイマー病の疑い」(SNAP)と診断されていた。1年以内の剖検で、TDP-43病理と海馬硬化症の存在が確認されLATE-NCであることが判明した。(B)91歳男性の認知症。アミロイドPETが陽性、タウPET陰性。MRIでは内側側頭葉の萎縮。このケースでは、初期のADNCとLATE-NCが共存していると推定されるが、このような病態の組み合わせは、特に高齢者の脳ではよく見られる。(Nelson PT. et al., Brain, 20119) {31039256}
専門医のための認知症テキスト
認知症関連ページ